更新了!多数据集合并及去除批次效应工具使用详解!
01
—
研究背景
批次效应表示样品在不同批次中处理和测量产生的试验期间记录与任何生物变异无关的技术差异。 高通量测序发展到现在,已有数十年,产生了海量的数据,数据的综合分析被认为是从基因组,转录组,蛋白组等组学数据集中提取最大有效信息的关键方法,目前这种方法更有利于研究人员从数据中发现和解决大的生物学问题[2-4]。对多个数据集进行合并是生信分析人员必备的技能之一。
然而,对数据进行合并并不是数据的简单叠加,早期的芯片测序试验中就可以观察到批次效应[5-6], 它的原因包括试验分析操作人员,时间、平台、实验室环境等[7]。 由于这些非生物因素的影响容易产生批次效应。很多情况下对不同组的数据进行整合时批次效应容易被忽略。尽管良好的试验设计可以减少批次效应, 但是很难彻底消除[8]。如果不消除批次效应,对下游的分析结果会产生很大的影响,例如做差异基因分析时,筛选出的差异基因究竟是由于批次效应导致的还是真实存在的差异。
目前消除批次效应的算法比较多,我们平台根据团队的项目经验和大量的文献调研选取了R软件包sva的combat函数进行去除批次效应,ComBat是基于经典贝叶斯的分析方法,运用已知的批次信息对高通量数据进行批次校正[9-10]。并进行尽心的打磨,配上丰富的结果展示图,可以直接插入sci写作中,为文章增色添彩。
话不多说,跟着小编一起去操作一番吧。
02
—
链接
http://sangerbox.com/Tool
03
—
使用方法
1.打开网址: http://sangerbox.com/Tool点击“多数据集合并及去除批次效应工具”即可进入分析界面。
2.准备数据: 将GSE118370、GSE27716、GSE18842三套数据来自同一注释平台(即GPL570)并下载到个人中心,如下图所示;
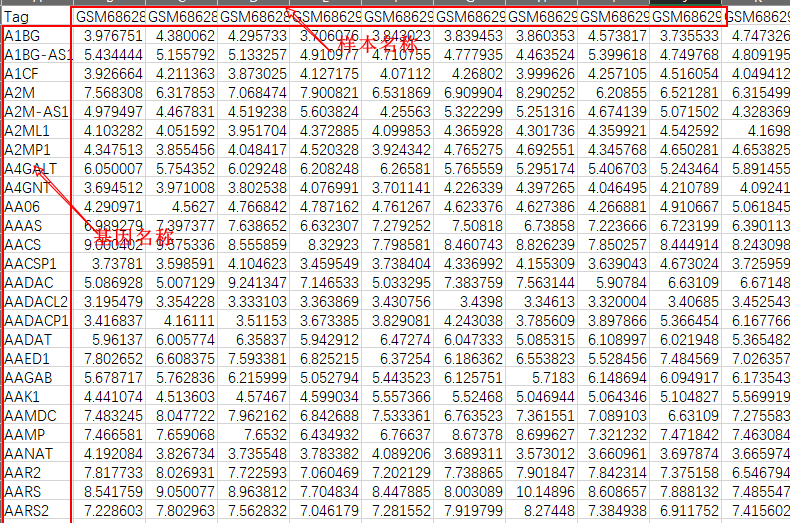
3.上面三套数据都有相同的格式 即行为基因名称,列为样本名称,如下图所示;
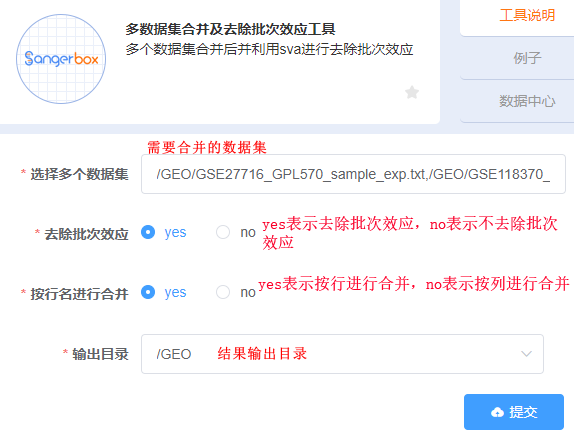
4.输入参数 如下图所示;
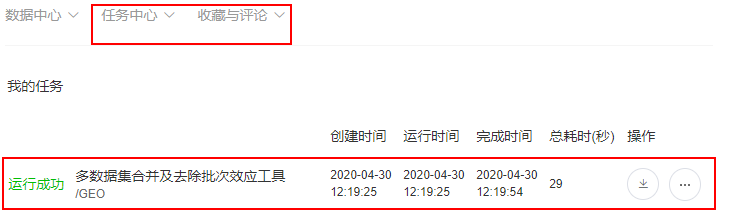
5.运行结果 判断结果是否完成,个人中心 任务中心查看任务是否完成,如下图所示。
04
—
结果展示
结果文件展示
结果展示
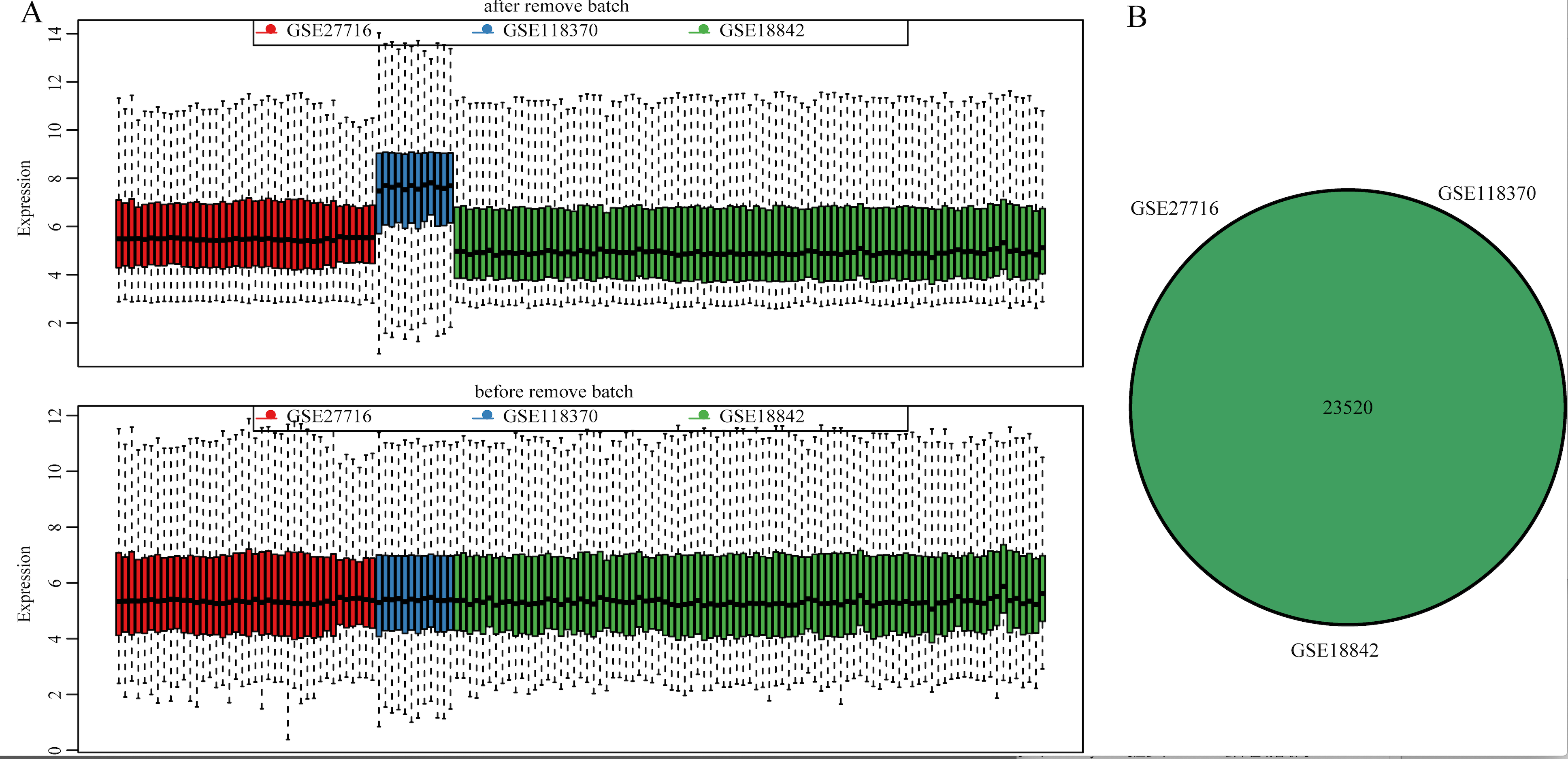
在结果文件中会生成两张图片,一张是不同数据集之间每个样本的箱线图,A图分别为去除批次效应之前的箱线图与去除批次效应之后的箱线图。B图为三个数据集列之间的venn图,因为上文选择的三个数据集是同一个注释平台的,探针数目一样,所以绘制成的venn图完全重合。
参考文献
[1] Rhodes, D., Chinnaiyan, A. Integrative analysis of the cancer transcriptome. Nat Genet 37, S31–S37 (2005)[2] LI Sa, ZHAO Yiqiang. Research progress on batch effect removal methods for gene expression data[J]. Journal of Nanjing Agricultural University, 2019, 42(3): 389-397.[3] Leek J T, Scharpf R B, Bravo H C, et al. Tackling the widespread and critical impact of batch effects in high-throughput data[J]. Nat Rev Genet, 2010, 11(10): 733-739.[4] Rhodes D R, Chinnaiyan A M. Integrative analysis of the cancer transcriptome[J]. Nat Genet, 2005, 37: S31-S37[5] Lander E S. Array of hope[J]. Nature Genetics, 1999, 21(51): 3-4.[6]Akey J M, Biswas S, Leek J T, et al. On the design and analysis of gene expression studies in human populations[J]. Nat Genet, 2007, 39(7): 807-809.[7]Fare T L, Coffey E M, Dai H, et al. Effects of atmospheric ozone on microarray data quality[J]. Anal Chem, 2003, 75(17): 4672-4675.[8] Chen C, Grennan K, Badner J, et al. Removing batch effects in analysis of expression microarray data:an evaluation of six batch adjustment methods[J]. PLoS One, 2011, 6(2): e17238.[9] Johnson W E, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods[J]. Biostatistics, 2007, 8(1): 118-127. DOI:10.1093/biostatistics/kxj037[10] Muller C, Schillert A, Rothemeier C, et al. Removing batch effects from longitudinal gene expression-quantile normalization plus comBat as best approach for microarray transcriptome data[J]. PLoS One, 2016, 11(6): e0156594.
具体指引详见:

- 发表于 2020-05-26 11:36
- 阅读 ( 14577 )
- 分类:软件工具
