文献解读(2)在鉴别肺腺癌患者与预后相关 immune signatures
点击"生信学霸"添加关注,我们一起进步
Systematic profiling of immune signatures identifies prognostic predictors in lung adenocarcinoma
在鉴别肺腺癌患者与预后相关 immune signatures
发表期刊:Cellular Oncology
发表日期:2020 May 28
影响因子:5.52
DOI: 10.1007/s13402-020-00515-7
01—
研究背景
世界范围内,肺癌是死亡率较高的癌症之一,其中肺腺癌(LUAD)是主要的分子亚型。尽管在过去十年中对LUAD的分子机制和临床管理有了很大的提高,如小分子抑制剂如酪氨酸激酶抑制剂(TKIs)可显着改善患者预后,但是通过靶向药物治疗会使患者产生异质性反应和耐药性。
尽管近年来,免疫治疗法在癌症治疗方面取得了很大的进展,如免疫抑制性PD-1,PDL1或CTLA4等免疫检查点抑制剂在抵抗多种恶性肿瘤取得显著的作用,但只有少数癌症患者从这些治疗中受益。
此外,获得性免疫药物治疗的耐药性确实存在,并且这种耐药性的机制尚不清楚,为了更好地指导临床免疫治疗,在这项研究中,作者识别了与免疫检查点抑制剂相关的生物标志物来预测患者的生存状况并阐明耐药性的分子机制。
02—材料和方法数据来源----------筛选差异基因数据集:GSE10072,GSE32863、GSE43458和TCGA RNA-seq表达谱数据训练集:GSE68465验证集:GSE31210,GSE50081和GSE72094
数据分析方法--------
原始数据预处理:对于GEO数据集采用分位数标准化和log2转化,对于TCGA数据集FPKM数据采用log2(FPKM+1)进行转化。差异基因分析:t-test并使用Benjamini-Hochberg(BH)对p值进行校正识别免疫相关基因:R 程序包RankAggreg差异基因富集分析:Cytoscape插件ClueGO却p<0.01识别预后基因:单因素Cox回归、一千次robsurv、多因素Cox回归、R程序包survival计算ROC计算免疫评分:R程序包CIBERSORT免疫组化分析:Human Protein Atlas (HPA) 网站
03
—
结果展示
1.筛选免疫相关的gene signatures从GEO数据库和TCGA数据库中获得LUAD癌症组织和正常组织mRNA表达数据,4个数据集的临床和病理特征统计如表1所示。表1. GEO数据集和TCGA数据集的临床信息统计
| Characteristics | GSE10072(T=58, N=49) | GSE32863(T=58, N=58) | GSE43458(T=80, N=30) | TCGA(T=56, N=56) | |
| Age | Median | 6845-87 | 7039-86 | NA | 6642-86 |
| Sex | Male | 35 (60.3%) | 13 (22.4%) | NA | 23 (41.1%) |
| Stage | I-II | 43 (74.1%) | 45 (77.6%) | NA | 41 (73.2%) |
| III-IV | 15 (25.9%) | 13 (22.4%) | NA | 15 (26.8%) |
T-肿瘤;N-正常从公共数据库中提取730个与免疫相关的基因,并从4个数据集中提取这些基因的表达谱,并用热图展示这些基因在四个LUAD表达谱中肿瘤样本和正常样本中的表达情况(图1A-D)。然后,使用t-test进行差异分析,根据阈值FDR<0.05和log2|FC|>1,在GSE10072数据集共筛选出43个免疫相关的差异基因,其中14个上调基因,29个下调基因;在GSE32863数据集共筛选出82个免疫相关的差异基因,其中15个上调基因67个下调基因;在GSE43458数据集共筛选出61个免疫相关的差异基因,其中15个上调基因46个下调基因;在TCGA数据集共筛选出124个免疫相关的差异基因,其中39个上调基因85个下调基因(图1E-H)。结果表明,与正常组织相比,免疫相关的基因在肿瘤组织中大多数基因表达量降低。进一步从四个数据集提取了205个与免疫相关的差异基因(图1I),发现这些下调基因的功能都参与了免疫反应过程,最后对这些基因进行富集分析,结果显示这些基因参与了T细胞增殖、白细胞迁移,巨噬细胞活化、NF-κB和TNF信号通路等生物学途径,这些表明了LUAD患者组织中失调的免疫相关基因是肿瘤免疫微环境变化的基础(图1K)。
注解:作者在这里筛选差异基因的方法用的是T检验,小伙伴们可以不常见,如果要重复他的结果可以使用limma。
2. 免疫组化反映(IHC)验证基因CD36在LUAD患者中表达的差异性在四个数据集中,使用R程序包RankAggreg删选前25个基因称为调节异常的免疫相关基因,阈值最小的5个基因分别是MARCO,CD36,LAMP3,CFD和PLA2G1B。查阅相关的文献,发现基因CD36在众多癌症起着重要的作用,所以选择基因CD36,并在四个数据集中的肿瘤和正常样品展示mRNA的表达水平(图2A)。接下来,作者在HPA数据集中搜索CD36基因在LUAD肿瘤样品和正常肺样品中的蛋白质表达水平,结果显示有三分之二的样本显示在正常组织中CD36高表达,在9.1%的样本中显示在肿瘤样本中高表达,其中有10个LUAD患者的样本没有检测到表水平(图2B),这与上面四个数据集mRNA的表达水平一致。此外作者还收集了43个配对的LUAD肿瘤和正常组织样本进行IHC分析,图2C显示了基因CD36没有表达、表达量低和表达量高的染色图像。这结果与上面的结果显示一致,CD36在正常组织比肿瘤组织中表达量中高(图2E-F)。
注解:作者在这里使用箱线图展示两组之间的差异,小伙伴们也可以使用小提琴图进行展示。
3.在LUAD数据集中构建与预后相关的免疫gene signature从GEO数据库中下载带有完整预后信息的4个数据集,分别为GSE68465、GSE72094、GSE50081和GSE31210他们的临床信息(见表2),其中GSE68465作为模型构建的训练集,其他三个作为模型构建的验证集。表2 构建基因分险模型训练集与验证集临床信息统计
| Characteristics | GSE68465(n=436) | GSE72094(n=381) | GSE50081(n=127) | GSE31210(n=226) | |
| Age | Median | 6533-87 | 7038-89 | 7040-86 | 6130-76 |
| Sex | Male | 219 (50.2%) | 166 (43.6%) | 65 (51.2%) | 105 (46.5%) |
| Stage | I-II | 369 (84.6%) | 311 (81.6%) | 127 (100%) | 226 (100%) |
| III-IV | 67 (15.4%) | 70 (18.4%) | 0 (0.0%) | 0 (0.0%) | |
| OS (days) | Median | 1414.05 | 832 | 1595.05 | 1744.5 |
| (range) | 60-6120 | 35-2077 | 32.85-3971.2 | 221-3863 |
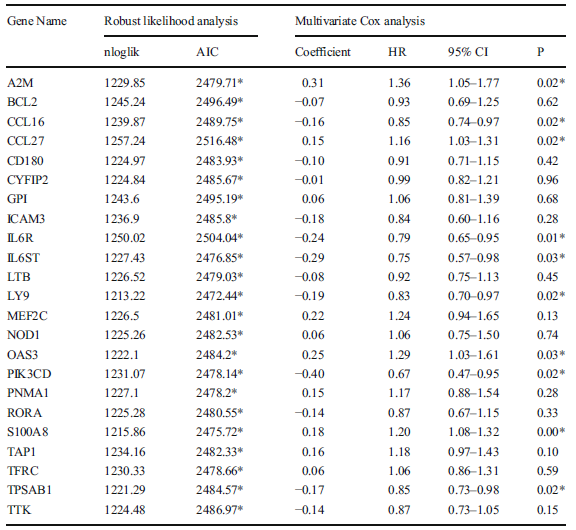
首先,使用训练集携带的生存数据,进行单变量Cox回归分析,共有132个免疫相关基因与患者的预后显著相关(p<0.05)。然后使用robust对132个免疫相关基因进一步进行降维分析,并根据cox模型中的似然函数选择23个基因做进一步分析。最后进行多因素cox回归分析,构建一个包含10个基因(A2M, CCL16, CCL27, IL6R, IL6ST, LY9, OAS3, PIK3CD, S100A8 and TPSAB1)的风险免疫基因模型,这10个基因中有6个基因(CCL16, IL6R, IL6ST, LY9, PIK3CD 和TPSAB1)HR小于1,为保护基因,4个基因(A2M, CCL27, OAS3 and S100A8)HR>1,为危险基因见表3。表3 训练集的robust生存分析及多元Cox分析结果
10个基因的KM曲线如图3B-K所示。尽管IL6ST, LY9 和TPSAB1的表达情况不能显著的(p>0.05)将患者分为高低分险两组(图3F/G/K),但是10个基因构建的分险预测模型能将患者分为高低分险两组,并且高风险组的生存时间比低风险短,且存在显著差异(图3L),且10个基因在2000天的ROC曲线的AUC超过了单个基因(图M)。
4.验证风险免疫基因模型模型为了评估风险免疫基因模型模型对其他肺腺癌数据也具有预测效率,作者使用三个独立的数据集来验证风险免疫基因模型模型的鲁棒性。结果显示,10个基因模型能显著将患者分为高低风险两组,且高风险组比低风险组存活时间短(图4A-C),且三个验证集在2000天具有很高AUC,且GSE72094,GSE31210和GSE50081数据集中,在2000天的AUC分别为0.69,0.72和0.74(图4D),说明10个免疫相关基因分险模型能够预测不同数据集患者的生存状态,就有广泛的预测性能。
5.免疫相关基因分险模型与临床信息的相关性作者进一步分析了免疫相关基因分析模型与临床变量的相关性(图5A),结果显示10个免疫风险基因表现出不同的表达水平。CCL27、OAS3和S100A8在肿瘤分化较低、有淋巴结转移、TNM分期晚期的男性患者中表达量较高。而IL6R、IL6ST、CCL16、A2M、TPSAB1、LY9和PIK3CD在肿瘤分化较好、无淋巴结转移、TNM分期早期的女性患者中表达量较高。然作者计算了免疫分险评分与患者的性别,肿瘤分化,肿瘤大小,淋巴细胞转移和TNM分期的相关性。结果显示:肿瘤分化较低、有淋巴结转移、TNM分期晚期的男性患者的分险得分要高于其他患者(图5B-C)。最后,将风险得分与临床变量因素进行多因素cox回归分析,结果显示免疫风险评分是LUAD患者的独立预后因素HR为2.46(图5D)。
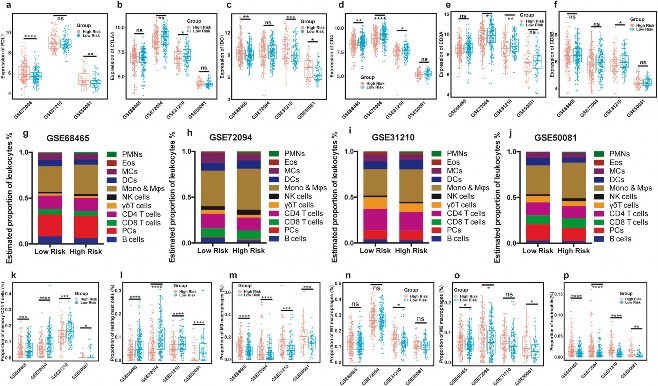
6.免疫相关基因分险模型与免疫的相关性最后,作者还分析免疫风险得分对免疫治疗的影响。首先,作者分析了与已经表明免疫治疗密切相关的基因,并根据风险得分的中值将患者分为高低风险两组,结果显示,在所有四个GEO数据集中,高风险组患者的PD-L1和IDO1的表达相对较高,但CTLA4、CD4、CD8A和CD8B的表达相对较低,GSE68465缺少PD-L1表达数据(图6A-F)。这些结果为预测患者对免疫治疗的反应提供了一些线索。众多研究表明,肿瘤微环境与肿瘤的发生发展密切相关,解开肿瘤复杂的免疫环境可能有助于揭示肿瘤难治性和免疫治疗耐药性的潜在机制。在这里,作者使用CIBERSORT工具计算每个样本的免疫评分,结果显示,高风险和低风险LUAD患者免疫微环境存在差异性,这些调节LUAD免疫微环境的潜在机制提供线索(图6G-P)。
04
—
结果图
图1.在肺腺癌患者的正常组织和肿瘤组织中筛选差异免疫相关基因
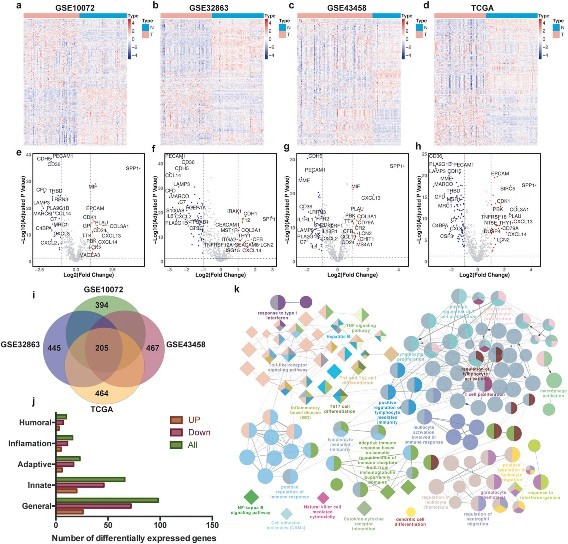
图2. 使用免疫组化(IHC)验证基因CD36 在 LUAD正常组织和肿瘤组织表达的差异性。 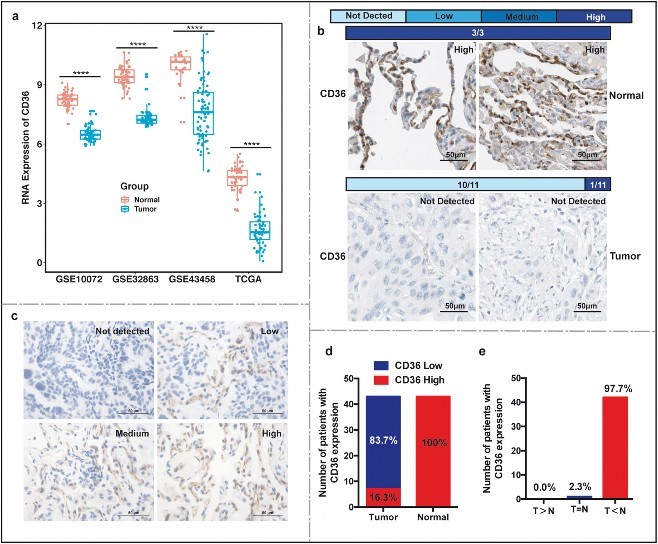
图3.鉴别与免疫相关的gene signature 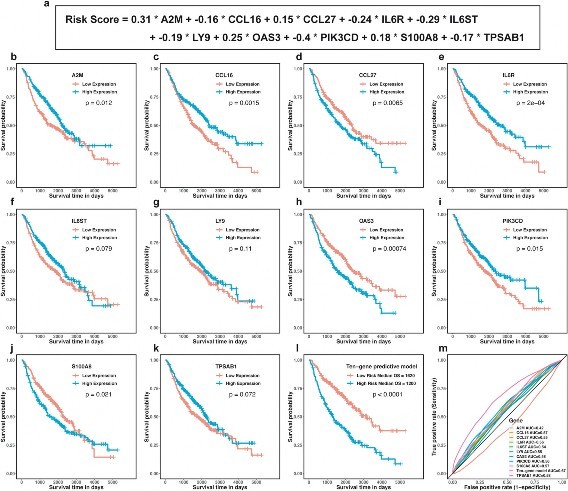
图4.外部验证集验证分险模型基因
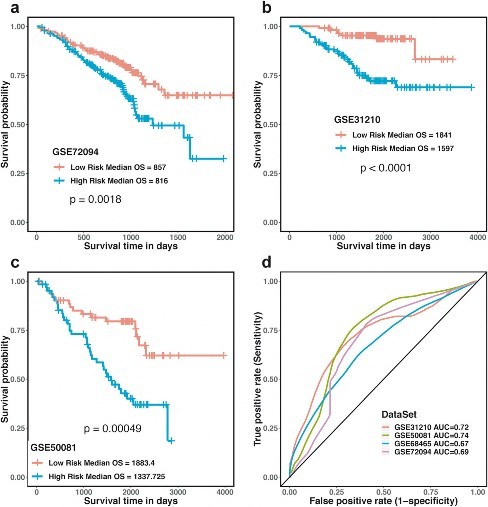
图5.基因分险评分与临床信息的关系 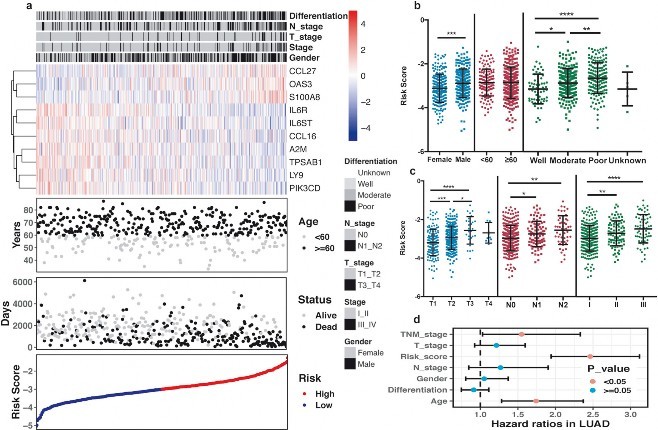
图6.基因分险评分与免疫评分的关系
05
—
结论
作者首先使用三套GEO数据加上TCGA数据,在正常组织和肿瘤组织中筛选与免疫相关的差异基因,并使用免疫组化进行验证,然后使用差异基因,在一套GEO数据中构建基因cox比例风险回归和rbsurv分析构建基因模型,并在三套外部数据集进行验证。

- 发表于 2020-06-15 15:47
- 阅读 ( 8179 )
- 分类:文献解读
