开源基因组浏览器JBrowse教程系列第二篇:使用拟南芥基因组演示怎么配置JBrowse
开源基因组浏览器JBrowse教程系列第二篇:使用拟南芥基因组演示怎么配置JBrowse
系统:Arch Linux
JBrowse版本:1.12.1
假设JBrowse安装目录为:/www/jb
假设下载保存路径为:/pub1/dl
假设JBrowse安装的服务器为:http://localhost:3003
本文是开源基因组浏览器JBrowse教程系列的第二篇,尚未部署好JBrowse的同学请移步第一篇安装篇
所谓基因组浏览器就是通过这个工具查看基因组,具体包括参考基因组序列,哪个地方是外显子、那个地方是内含子等等功能。参考基因组就是一个fasta文件,哪个地方是外显子、哪个地方是内含子这些信息称之为特征,一般情况下NCBI、ENSEMBL等数据库都会提供GFF3格式的基因组特征文件,关于GFF3格式的说明请参考我的另外一篇文章GFF3格式说明。
以下以拟南芥为例演示怎么使用JBrowse。
第一步:准备数据
下载拟南芥基因组:
$ wget ftp://ftp.ensemblgenomes.org/pub/release-36/plants/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
下载拟南芥基因组特征文件:
$ wget ftp://ftp.ensemblgenomes.org/pub/release-36/plants/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.36.abinitio.gff3.gz
解压:
$ gzip -d Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz $ gzip -d Arabidopsis_thaliana.TAIR10.36.abinitio.gff3.gz
第二步:格式化参考基因组
JBrowse安装目录下的bin目录提供了很多方便使用的perl脚本,例如:
$ cd /www/jb $ ls bin add-bam-track.pl add-json.pl bam-to-json.pl cpanm flatfile-to-json.pl jbdoc maker2jbrowse prepare-refseqs.pl ucsc-to-json.pl add-bw-track.pl add-track-json.pl biodb-to-json.pl draw-basepair-track.pl generate-names.pl json2conf.pl new-plugin.pl remove-track.pl wig-to-json.pl
其中bin/prepare-refseqs.pl是用来格式化参考基因组序列的。
使用方法为:
$ prepare-refseqs.pl --fasta <file1>
即:
$ bin/prepare-refseqs.pl --fasta /pub1/dl/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa
这时你看一下data目录,发现目录下多了一些文件:
$ tree -L 2 data data ├── seq │ ├── 1ad │ ├── 536 │ ├── 6dd │ ├── 83d │ ├── 84b │ ├── ac0 │ ├── f3b │ └── refSeqs.json ├── trackList.json └── tracks.conf
其中seq目录下的1ad、536等等目录都是存放格式化好的参考基因组文件的。
第三步:格式化特征文件
格式化特征文件需要用到bin/flatfile-to-json.pl这个脚本,使用方式为:
$ bin/flatfile-to-json.pl (--gff <GFF3 file> | --bed <BED file> | --gbk <GenBank file>) --trackLabel <track identifier>
也就是说你这个特征文件可以是GFF3、BED、GBK三种中的一种(熟悉Linux命令行的同学应该能够很轻易地看懂这个说明),另外必须提供trackLabel这个参数,来指定这个track的ID,这个参数会作为名字显示在基因组浏览器的左侧的tracks那一列。
即:
$ bin/flatfile-to-json.pl --gff /pub/dl/Arabidopsis_thaliana.TAIR10.36.abinitio.gff3 --trackLabel 'GFF3 Annotations'
运行完毕,再打开data/trackList.json你就会发现多了一些东西:
{
"tracks" : [
{
"seqType" : "dna",
"key" : "Reference sequence",
"storeClass" : "JBrowse/Store/Sequence/StaticChunked",
"chunkSize" : 20000,
"urlTemplate" : "seq/{refseq_dirpath}/{refseq}-",
"label" : "DNA",
"type" : "SequenceTrack",
"category" : "Reference sequence"
},
{
"style" : {
"className" : "feature"
},
"key" : "GFF3 Annotations",
"storeClass" : "JBrowse/Store/SeqFeature/NCList",
"trackType" : null,
"urlTemplate" : "tracks/GFF3 Annotations/{refseq}/trackData.json",
"compress" : 0,
"type" : "FeatureTrack",
"label" : "GFF3 Annotations"
}
],
"formatVersion" : 1
}
这个文件是当前的所有的track,以JSON文件存储,其实就是一个文本而已,不过这个文本对程序是友好的,仔细阅读你就会发现有两个track,再看看不就是我刚刚格式化的参考基因组和特征文件嘛!
这个时候一般你打开浏览器输入http://localhost:3003就可以访问了,但是有一个隐患。JBrowse默认是支持多个基因组的,而且默认bin目录下的各种脚本的输出路径都是data目录,那如果我下次又想弄一个基因组,经过上面两步也放到了data目录,那岂不是很混乱?JBrowse已经为我们想好了解决方案,你只需要把现在的data目录改一下名字就行了:
$ mv data Arabidopsis_thaliana
下次再把另外一个基因组放进来时,bin目录下的脚本又会默认放到data目录,就不会发生混乱啦!
这时你访问http://localhost:3003就会发现这是一个错误页,因为http://localhost:3003不加参数的话默认就是访问data目录的数据,现在你把data目录重命名当然不存在啦。现在想访问刚刚的拟南芥数据可以给这个URL加上get参数,即:http://localhost:3003/?data=Arabidopsis_thaliana,也就是把data=后面的字符换成刚刚修改后的文件夹的名字。
另外,如果你把JBrowse部署在内网,却想让外网访问到,而部署JBrowse的服务器又在防火墙后面的话,请一定要记得把jbrowse.conf加入到防火墙白名单,因为防火墙默认会把这种.conf结尾的文件屏蔽。
最后看下效果:
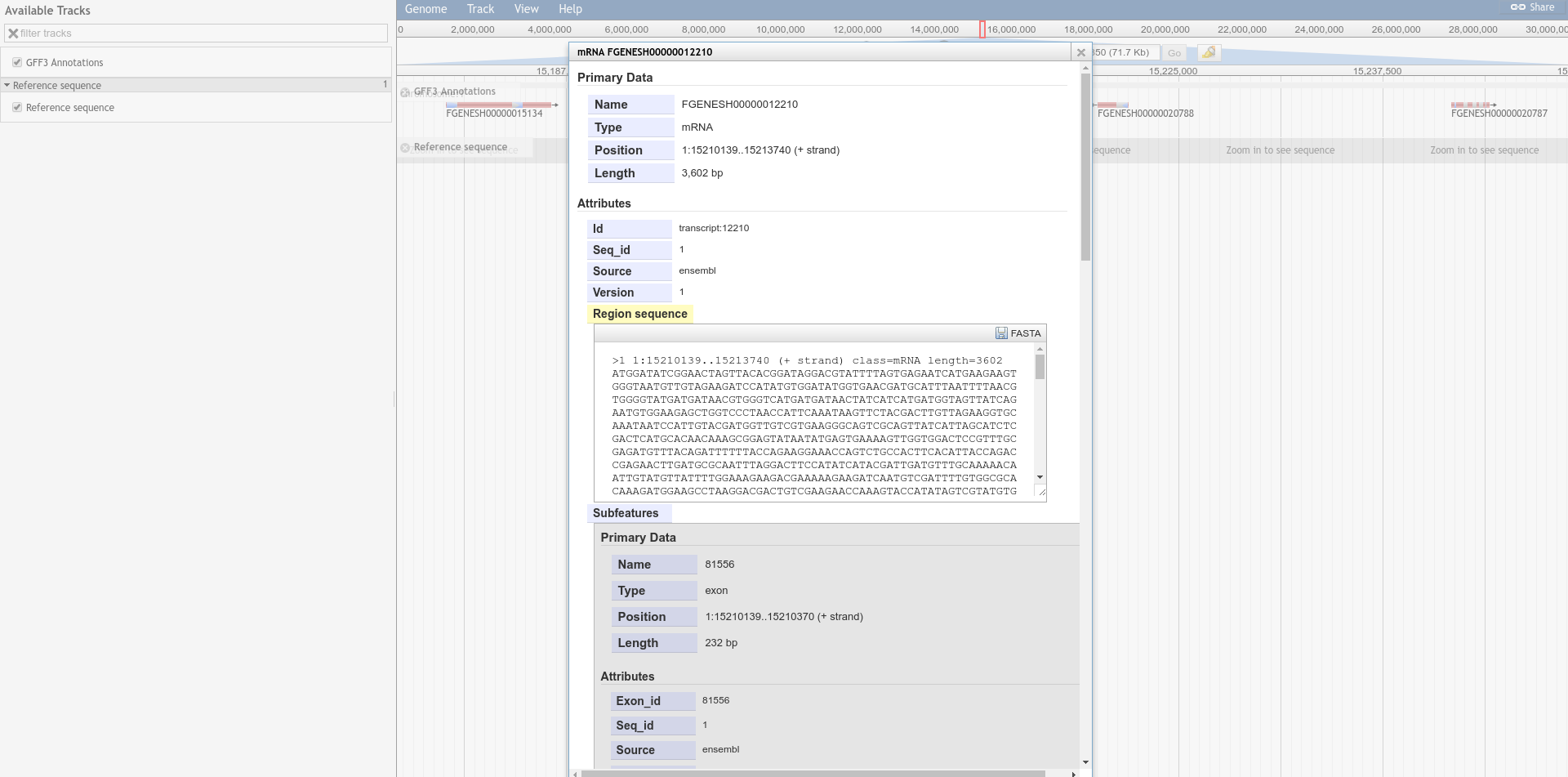
- 发表于 2017-08-09 17:31
- 阅读 ( 8286 )
- 分类:软件工具
