Vcf文件怎么看?
什么是Vcf文件?
Vcf,即variant call format,是用来描述SNP、INDEL、SV等变异结果的一种文件格式。我们在使用GATK或者samtools做SNP calling时,得到的就是vcf格式的文件。那么这个文件该怎么看呢?
例子:
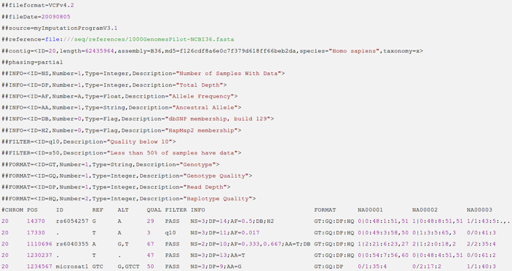
VCF文件是文本文件,任意一款文本编辑器都能打开,从上面的例子上看,可以分为以两个“#”号开头的注释说明部分和剩下的主体部分。注释说明部分主要为主体部分出现的一些tag的说明,以key=value形式记录信息,如INFO、FORMAT中的DP、AP、GT等。而主体部分包括一行以“#”号开头的表头信息和其他的数据行,每个数据行记录一个variant的信息,我们做分析所需要的数据就在这里。主体部分每列的信息:
CHROM和POS:变异位点所在的染色体名称和位置,从1开始计数,如果是INDEL的话,位置是该INDEL第一个碱基的位置。
ID:variant的id。比如call出来的SNP在dbSNP数据库中存在,这里就会显示相应的rs号(当然前提是已经和dbSNP数据库做了比较)。
REF和ALT:参考序列的碱基和突变后的碱基。如果有多种不同于参考序列的基因型,在ALT列使用“,”隔开。如变异位点在参考基因组上的碱基为“G”,样品上突变后的基因型为“A”,则REF列为“G”,ALT列为“A”;如果突变后的碱基有多个如A和C,则ALT可以表示为“A,C”。这里需要注意ALT是针对这个变异位点而言,不针对特定样品。
QUAL:Phred格式(Phred_scaled)的质量值,表示在该位点存在variant的可能性,值越高,则variant的可能性越大。计算方法:Phred值= -10 * log (1-p), p为variant存在的概率。通过计算公式可以看出值为10的表示错误概率为0.1,该位点为variant的概率为90%。
FILTER:理想情况下,QUAL这个值应该是用所有的错误模型算出来的,这个值就可以代表正确的变异位点了,但是事实是做不到的。因此,还需要对原始变异位点做进一步的过滤。无论你用什么方法对变异位点进行过滤,过滤完了之后,在FILTER一栏都会留下过滤记录,如果是通过了过滤标准,那么这些通过标准的好的变异位点的FILTER一栏就会注释一个PASS,如果没有通过过滤,就会在FILTER这一栏提示除了PASS的其他信息。如果这一栏是一个“.”的话,就说明没有进行过任何过滤。
INFO:这一列是variant的详细信息,格式以tag=value形式记录,而tag的说明一般包含在文件开头的注释说明部分。
FORMAT和NA00001(NA00002...):FORMAT这列规定了后边样品每列的格式,NA00001(NA00002...)等各列是对应每个样品在这个variant的信息。我们如果要看每个样品的基因型信息,就需要看这几列了。
那样品的基因型怎么看呢?
每个样品的基因型格式都是一样的,通过FORMAT列来定义,如FORMAT列为GT:GQ:DP,样品列为0/1:35:4,则相对应的样品的GT为0/1,GQ为35,DP为4。那么这些又表示什么呢?我们解释一下经常出现在FORMAT中的tag,你就明白了:
GT:样本的基因型(genotype)。两个数字中间用‘/’分开,这两个数字表示双倍体的sample的基因型。0 表示样品中有ref的allele;1表示样品中variant的allele;2表示有第二个variant的allele。因此:0/0表示sample中该位点为纯合的,和ref一致;0/1 表示sample中该位点为杂合的,有ref和variant两个基因型;1/1 表示sample中该位点为纯合的,和variant一致。如果REF是A,ALT是C的话,则0/0就表示AA,0/1表示AC,1/1表示CC。
AD和DP:AD(Allele Depth)为sample中每一种allele的reads覆盖度,在diploid中则是用逗号分割的两个值,前者对应ref基因型,后者对应variant基因型;DP(Depth)为sample中该位点的覆盖度。
GQ:基因型的质量值(Genotype Quality)。Phred格式(Phred_scaled)的质量值,表示在该位点该基因型存在的可能性;该值越高,则Genotype的可能性越大;计算方法:Phred值= -10 * log (1-p) p为基因型存在的概率。
PL:指定的三种基因型的质量值(provieds the likelihoods of the given genotypes)。这三种指定的基因型为(0/0,0/1,1/1),这三种基因型的概率总和为1。和之前不一致,该值越大,表明为该种基因型的可能性越小。Phred值= -10 * log (p) p为基因型存在的概率。
了解了之后,我们就知道,每个样品的基因型是通过GT部分的内容确定的,而深度信息可以通过AD或DP来看。再结合CHROM、POS、REF、ALT等,就可以知道每个variant的具体信息了。
以上就是整个vcf文件内容了,了解了上面的信息,就可以根据自己的需要对vcf文件进行查看、处理。最后给大家推荐一款vcf文件的处理工具vcftools,它专门针对vcf文件进行处理,包含过滤、提取、合并、排序、统计等各种功能,软件地址:
https://vcftools.github.io/index.html。
- 发表于 2017-04-07 10:16
- 阅读 ( 18035 )
- 分类:软件工具
