用小提琴图来可视化基因差异表达之美化过程
为了表示基因在样本中的差异,对于许多个基因的话用火山图感觉挺高大上的,那么对于少数几个基因的话一般用什么来展示其表达差异呢,有很多种办法,比如箱线图,柱状图等等,今天主要用小提琴图来可视化几个基因的差异表达。
为了表示基因在样本中的差异,对于许多个基因的话用火山图感觉挺高大上的,那么对于少数几个基因的话一般用什么来展示其表达差异呢,有很多种办法,比如箱线图,柱状图等等,今天主要用小提琴图来可视化几个基因的差异表达。
首先安装R包
source("https://bioconductor.org/biocLite.R")
biocLite('vioplot')
然后我的数据是一个矩阵,行为样本列为基因如:

已经知道癌症样本和正常样本的索引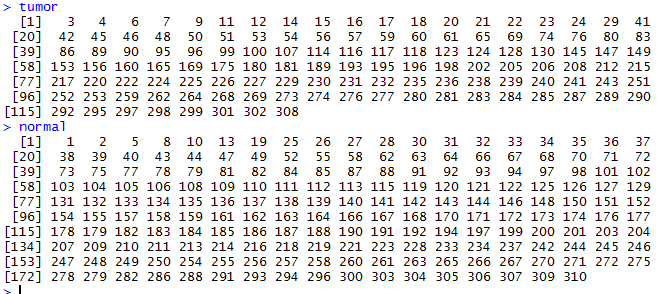
那么绘制小提琴图的简单办法如下:
vioplot(sigData[tumor,1]
,sigData[normal,1]
,sigData[tumor,2]
,sigData[normal,2]
,sigData[tumor,3]
,sigData[normal,3]
,sigData[tumor,4]
,sigData[normal,4]
,names=rep(c("Tumor","Normal"),4),col="red")
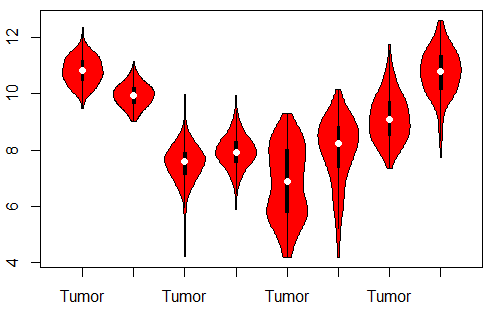
从图中可以看出这四个基因其实在癌与癌旁是有差异的,但是好丑,很显然横轴要标上基因,每一个基因的癌与癌旁近一点,同时标记上p值最好了
那么怎么调整呢
首先需要调整的小提琴图的间距,让其看起来像四组,而不是八组,我们机智的设计了x轴,如代码所示:
x=c(1:5)
y=c(1:5)
plot(x, y, xlim=c(0,10),xlab='',ylab='Gene Expression'
,main=''
, ylim=c(min(sigData),max(sigData)+1),pch=21,col='white',xaxt="n")
for(i in 1:4){
basal=sigData[tumor,i]
noBasal=sigData[normal,i]
vioplot(add = T,basal,at=3*(i-1),lty=1)
vioplot(add = T,noBasal,at=3*(i-1)+1,lty=1)
}
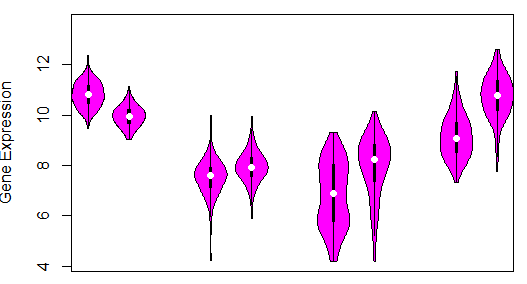 从代码中可以看出我们将x轴设计成了0-10,然后再0,1,2,3,5,6,8,9处画上小提琴,这样就可以看出四组数据了,顺便将y轴的label加上
从代码中可以看出我们将x轴设计成了0-10,然后再0,1,2,3,5,6,8,9处画上小提琴,这样就可以看出四组数据了,顺便将y轴的label加上
但是这个还是很丑,我们想将tumor和normal用颜色区分一下下,怎么办,如代码:
x=c(1:5)
y=c(1:5)
plot(x, y, xlim=c(0,10),xlab='',ylab='Gene Expression'
,main=''
, ylim=c(min(sigData),max(sigData)+1),pch=21,col='white',xaxt="n")
for(i in 1:4){
basal=sigData[tumor,i]
noBasal=sigData[normal,i]
vioplot(add = T,col = 'blue',basal,at=3*(i-1),lty=1)
vioplot(add = T,col = 'red',noBasal,at=3*(i-1)+1,lty=1)
}
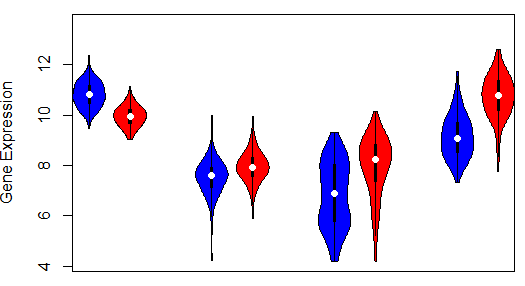 如咱们所愿,颜色变了,但是p值木有加上,还是挺遗憾的,咱们使用‘Mann-Whitney’ test来检验差异显著性,然后把p值在图中,做法如下:
如咱们所愿,颜色变了,但是p值木有加上,还是挺遗憾的,咱们使用‘Mann-Whitney’ test来检验差异显著性,然后把p值在图中,做法如下:
x=c(1:5)
y=c(1:5)
plot(x, y, xlim=c(0,10),xlab='',ylab='Gene Expression'
,main=''
, ylim=c(min(sigData),max(sigData)+1),pch=21,col='white',xaxt="n")
for(i in 1:4){
basal=sigData[tumor,i]
noBasal=sigData[normal,i]
vioplot(add = T,col = 'blue',basal,at=3*(i-1),lty=1)
vioplot(add = T,col = 'red',noBasal,at=3*(i-1)+1,lty=1)
p=round(wilcox.test(basal,noBasal)$p.value,3)
mx=max(c(basal,noBasal))
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))
text(x=3*(i-1)+0.5,y=mx+0.5,labels=ifelse(p==0,paste0("p<0.001"),paste0("p<",p)),cex = 0.5)
}
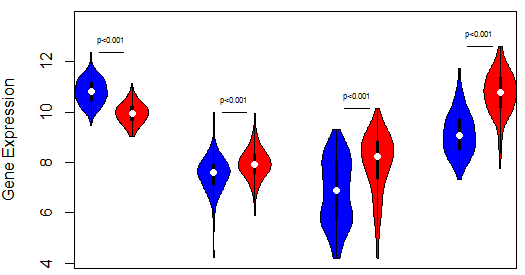
从图中可以看出p值已经加上去了,对比之前的代码主要多了四行代码
p=round(wilcox.test(basal,noBasal)$p.value,3)#用来检验基因的差异显著性p值,并去3位有效数字
mx=max(c(basal,noBasal))#找到新画上去的两个小提琴的最高的位置,为画上p值的位置坐准备
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))#在画p值之前先画一条横跨在癌与癌旁的两个小提琴之间的横线,注意那个0.8,两个小提琴直接的间距是1,那么0.8表达离第二个小提琴的位置还差0.2
text(x=3*(i-1)+0.5,y=mx+0.5,labels=ifelse(p==0,paste0("p<0.001"),paste0("p<",p)),cex = 0.5)#画上p值的文本啦,x,y都是图中位置,仔细体会,label就是文本,cex设成0.5表示原来的50%
p值也画上去了,但是x轴一直没有,最后咱们来绘制x轴,首先分析一下八个小提琴的位置咱们是知道的,分别在0,1,2,3,5,6,8,9位置上,所以咱们先绘制八个癌与癌旁的小label
x=c(1:5)
y=c(1:5)
plot(x, y, xlim=c(0,10),xlab='',ylab='Gene Expression'
,main=''
, ylim=c(min(sigData),max(sigData)+1),pch=21,col='white',xaxt="n")
for(i in 1:4){
basal=sigData[tumor,i]
noBasal=sigData[normal,i]
vioplot(add = T,col = 'blue',basal,at=3*(i-1),lty=1)
vioplot(add = T,col = 'red',noBasal,at=3*(i-1)+1,lty=1)
p=round(wilcox.test(basal,noBasal)$p.value,3)
mx=max(c(basal,noBasal))
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))
text(x=3*(i-1)+0.5,y=mx+0.5,labels=ifelse(p==0,paste0("p<0.001"),paste0("p<",p)),cex = 0.5)
}
axis(side=1, at = c(0,1,3,4,6,7,9,10), labels = F, tick = TRUE)
text(c(0,1,3,4,6,7,9,10),rep(par("usr")[3]-0.7,8),xpd = NA,cex = 0.8,labels=rep(c('Basal','non-Basal'),4),srt = 45,adj=1)
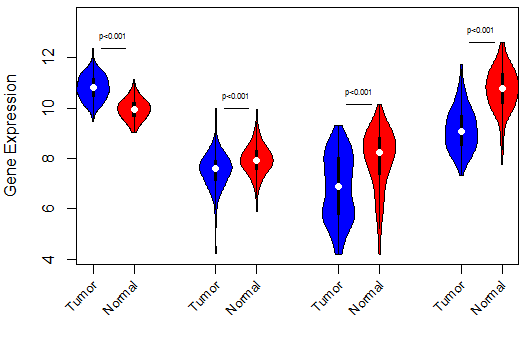 对比代码咱们加了两行
对比代码咱们加了两行axis(side=1, at = c(0,1,3,4,6,7,9,10), labels = F, tick = TRUE)#表示坐标抽啦,side=1表示横轴,at= 表示八个位置,labels=F表示不绘制label,tick=T表示显示那个小竖杆
text(c(0,1,3,4,6,7,9,10),rep(par("usr")[3]-0.7,8),xpd = NA,cex = 0.8,labels=rep(c('Basal','non-Basal'),4),srt = 45,adj=1)#绘制那些文字啦,srt表示倾斜角度
#注意:rep(par("usr")[3]-0.7,8)很重要,他直接表示了垂直方向的位置,尤其是那个0.7,可以试着改大改小,好好体会一下
最好要把基因给标上去嘛,代码如下:
x=c(1:5)
y=c(1:5)
plot(x, y, xlim=c(0,10),xlab='',ylab='Gene Expression'
,main=''
, ylim=c(min(sigData),max(sigData)+1),pch=21,col='white',xaxt="n")
for(i in 1:4){
basal=sigData[tumor,i]
noBasal=sigData[normal,i]
vioplot(add = T,col = 'blue',basal,at=3*(i-1),lty=1)
vioplot(add = T,col = 'red',noBasal,at=3*(i-1)+1,lty=1)
p=round(wilcox.test(basal,noBasal)$p.value,3)
mx=max(c(basal,noBasal))
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))
text(x=3*(i-1)+0.5,y=mx+0.5,labels=ifelse(p==0,paste0("p<0.001"),paste0("p<",p)),cex = 0.5)
}
axis(side=1, at = c(0,1,3,4,6,7,9,10), labels = F, tick = TRUE)
text(c(0,1,3,4,6,7,9,10),rep(par("usr")[3]-0.7,8),xpd = NA,cex = 0.8,labels=rep(c('Tumor','Normal'),4),srt = 45,adj=1)
text(c(0.5,3.5,6.5,9.5),rep(par("usr")[3]-3,8),xpd = NA,labels=colnames(sigData),cex = 0.8)
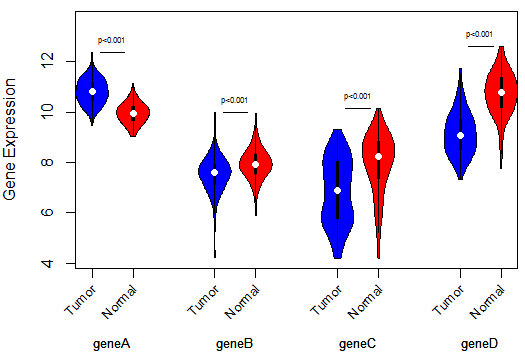
就这样完成了
- 发表于 2017-07-03 21:09
- 阅读 ( 22898 )
- 分类:软件工具
