谁来调控我感兴趣的DNA?100%可行的全面解决方案
本文关心的是直接调控,即哪个蛋白/RNA直接结合我感兴趣的DNA。研究哪个蛋白质结合某段DNA,有两种方法screen:
Plan A:大量ChIP-seq公共数据
Plan B:motif分析预测
本文关心的是直接调控,即哪个蛋白/RNA直接结合我感兴趣的DNA。研究哪个蛋白质结合某段DNA,有两种方法screen:
- Plan A:大量ChIP-seq公共数据
- Plan B:motif分析预测
研究哪个RNA直接结合某段DNA,可以通过大量的ChiP-seq数据分析解决。目前人的ChiRP-seq数据不足100套,随着数据的积累,也会像下面介绍的Plan A一样,找到结合DNA的RNA。
本文最后会介绍一下低通量的实验验证方法。
Plan A:基于大量ChIP-seq公共数据
目前全世界已发表人和小鼠的2万多套ChIP-seq数据,包含800多个TF,把这些ChIP-seq数据放在一起,就能看到基因组的每个位置都结合了哪些TF。
进入UCSC 能查询到ENCODE产生的167个TF和组蛋白修饰的ChIP-seq数据。


找到Regulation,点击ENC TF binding...,show,refresh
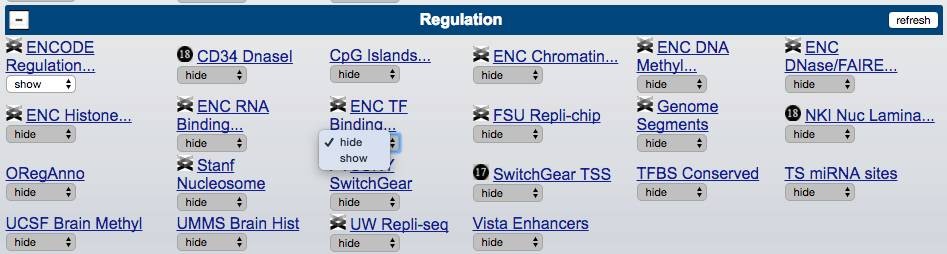
refresh后,就能看到这段DNA范围内有结合信号peak的TF,例如NFYA、E2F1等
左侧依次是细胞系的名字和TF的名字

如果不巧,是167个以外的某个TF对我的这段DNA起了关键的调控作用,不就看不到了吗?说好的800多个TF的20000套ChIP-seq数据呢?如何查看呢?
这就要用到上篇提到的CistromeDB。
CistromeDB提供了批量下载功能,http://cistrome.org/db/#/
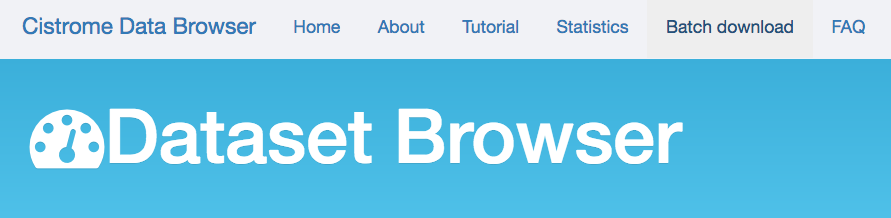
点击右上角的“Batch download”,填写课题组信息,勾选要下载的数据类型
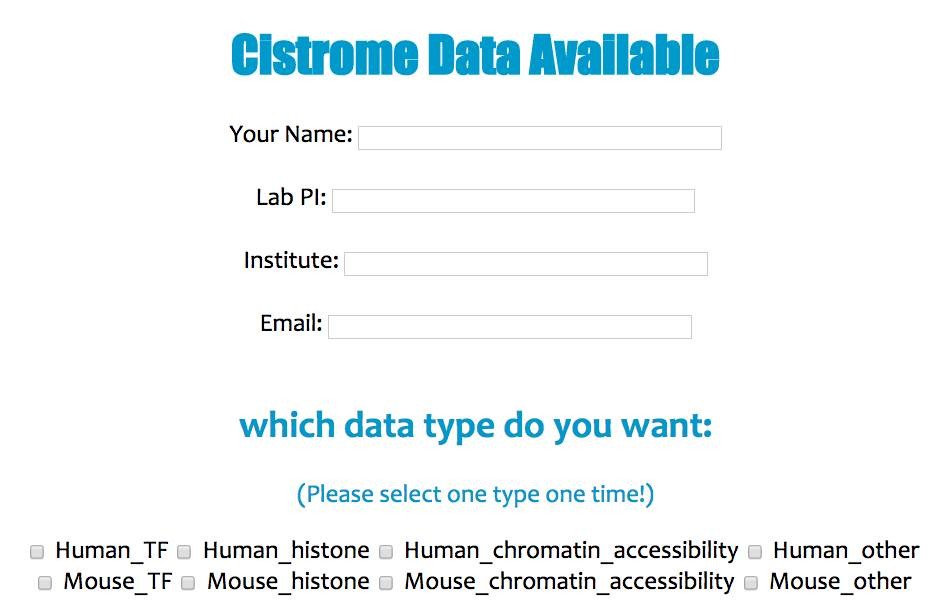
承诺提交的信息正确,不会把下载到的数据交给别人,发表文章的时候引用该论文。输入校验码,点击最下面的按钮,就开始下载了。
用bedtools找出感兴趣的基因附近有结合信号peak的ChIP-seq数据,对应到TF名字,就推测出哪些TF结合了感兴趣的基因。bedtools的用法满天飞,小哈在这里不啰嗦。
该方法的优点是,找到的TF跟DNA关系是有in vivo实验证据的。
缺点是,基因的转录调控有着组织特异性,在这套ChIP-seq数据的细胞类型和处理条件下不结合,不代表你关心的细胞类型或处理条件下也不结合,有可能真就能结合呢!反之亦然。
Plan B:基于motif预测
通过motif预测DNA上可能会有哪些转录因子结合。每个转录因子都有一个DNA结合结构域(DBD),喜欢结合在特定DNA序列上,也就是motif。
如果我感兴趣的基因上游DNA有某个TF的motif,那么该TF就有可能结合这段DNA,从而调控下游基因表达。
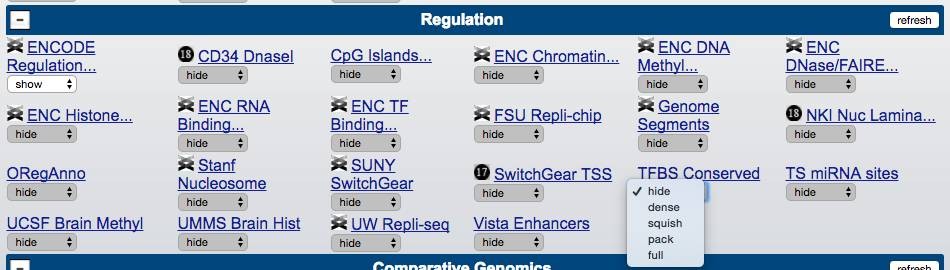
进入UCSC,https://genome.ucsc.edu/cgi-bin/hgGateway,找到Regulation,点击TFBS Conserved,full,refresh
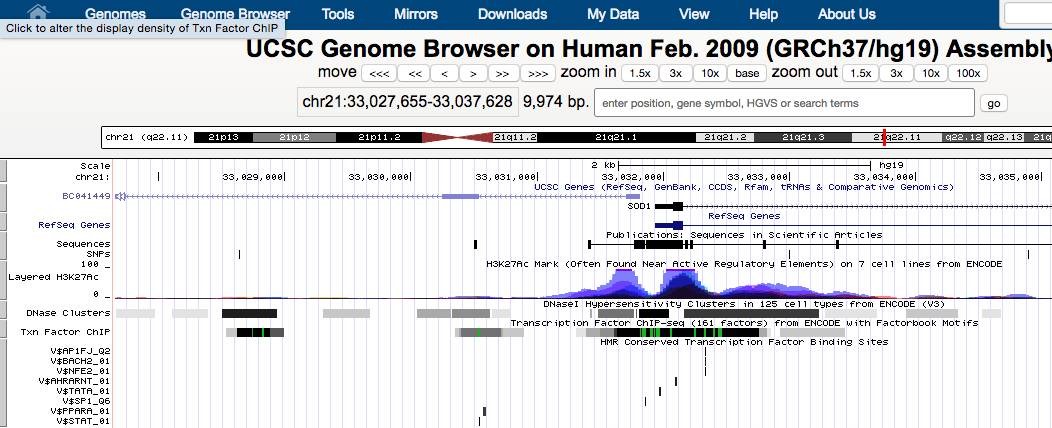
点击名字,出现motif信息
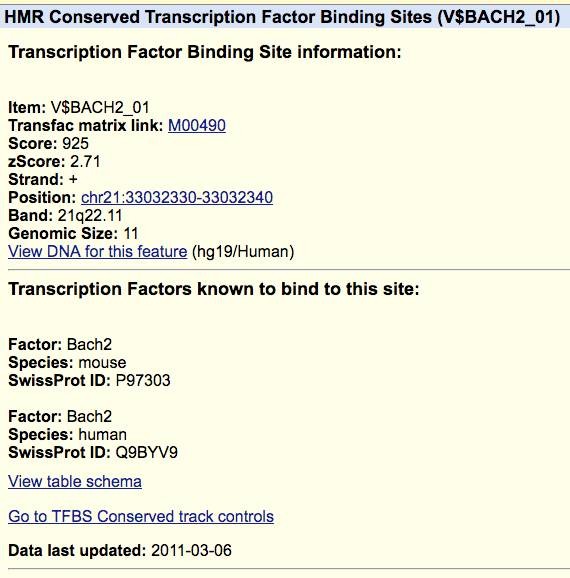
该方法的缺点是,就算在DNA序列上找到了TF对应的motif,并且用EMSA实验验证是阳性,该TF不一定真的就能in vivo结合这段DNA。这起码提供了一条线索,让你有迹可循,看到了某个感兴趣的TF的motif,就做个ChIP-qPCR验证一下吧!
低通量实验验证蛋白质-DNA结合
ChIP-qPCR,验证细胞内真实存在的某个蛋白质与某段DNA的结合情况。如果蛋白质跟这段DNA结合,加蛋白质的抗体就能拉下这段DNA,对照组不加抗体。在这段DNA上设计引物,做qPCR,ChIP样品里该段DNA的扩增产物会远高于对照。缺点是,ChIP实验对技术要求高,不一定有好用的抗体。
EMSA,又叫Gel shift assay,凝胶迁移滞后实验。跑电泳时,DNA/RNA自己像老鼠,跑得快;如果DNA/RNA背上蛋白质,像大象,跑的慢。优点是检测对象是DNA,不需要蛋白质的抗体,缺点是不能代表细胞内的真实结合情况。
DNA footprinting,裸露的DNA被DNase随机切成长短不一的片段;如果某段DNA上结合了蛋白质,就不会被DNase切到。跑胶,理想状态是对照组DNA样品在胶里是均匀分布的;蛋白质保护DNA不被酶切,导致结合了蛋白质的样品中间会空一块。理想很丰满,现实是DNA上已经结合的调控蛋白形成DHS,以及酶本身的bias,导致对照组就不是均匀分布。
另外,RNA footprinting,研究蛋白质与RNA的结合,原理类似于DNA footprinting,发表在2014年的Genome Biology上。缺点是技术难度大。

- 发表于 2018-11-16 20:46
- 阅读 ( 5193 )
- 分类:转录组学
