TCGA简易下载工具V16版升级详细教程
前言:
关于老版本:从TCGA最早的V6到现在的V16,我们一直在努力更新,让他变得更好,当然现在的老版本V8及以下的都不能正常使用了,这是由于GDC API做了限制,一次最多只能查询10000个样本的数据,而老版本查询时一次查询多于该限制阈值,所以老版本的都不能用了,会一直显示超时!!!
关于ID转换:老版本的ID转换无法做到及时更新,所以把这个功能单独拿出来做成了另外一个插件,简易ID转换工具,故两者可以配合使用,即可达到ID转换的目的.
关于正常样本和肿瘤样本分开:因为涉及到大的矩阵操作经常会内存不够,所以新版本中取消了自定义合并的功能,目前使用可以有两种方式达到目的,一种是分开下载合并成两个矩阵后再进行合并,一种是利用:简易矩阵操作工具,来自定义你的大矩阵的列的排序和行的排序方式。
配了鸡汤也配了勺子,喝不喝下去你开心就好。
工具的下载原理是利用GDC API,感兴趣的可以去看一看。
这是一个极简的下载工具,从盒子打开之后(打不开的看这里),他长这个样子:
对,就是这样,看着啥都没有!!!
其实他是在检索,大约过一会儿(因你的网络而定)就会长这个样子:
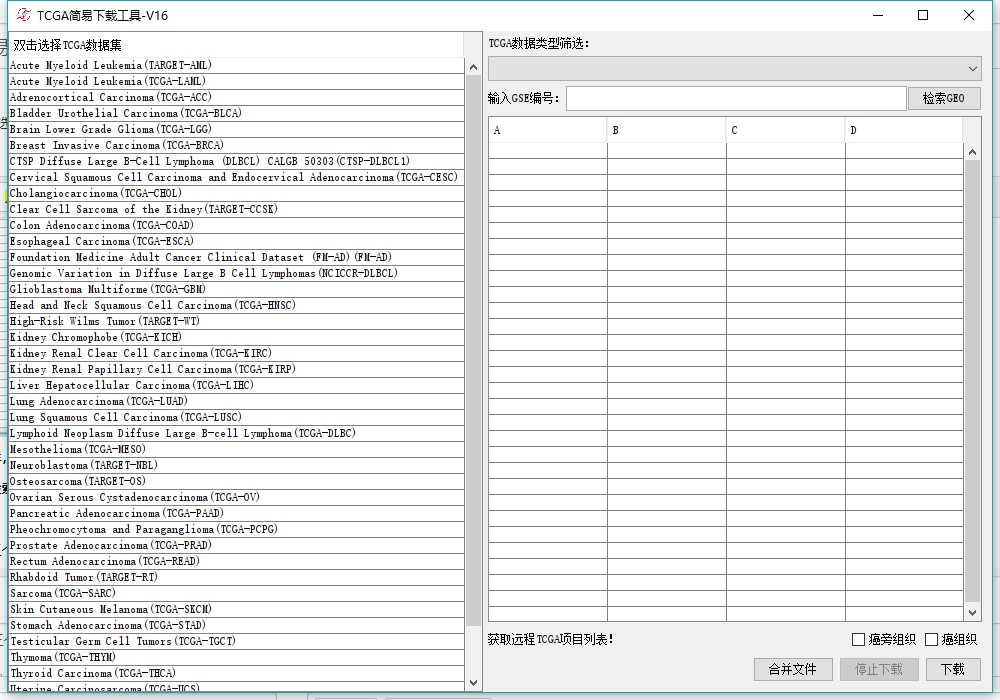 左侧就是检索出来可以下载的TCGA的三十多种肿瘤,你可以双击任何的一个肿瘤便可以进行下一步操作,比如双击选择胃癌:
左侧就是检索出来可以下载的TCGA的三十多种肿瘤,你可以双击任何的一个肿瘤便可以进行下一步操作,比如双击选择胃癌:
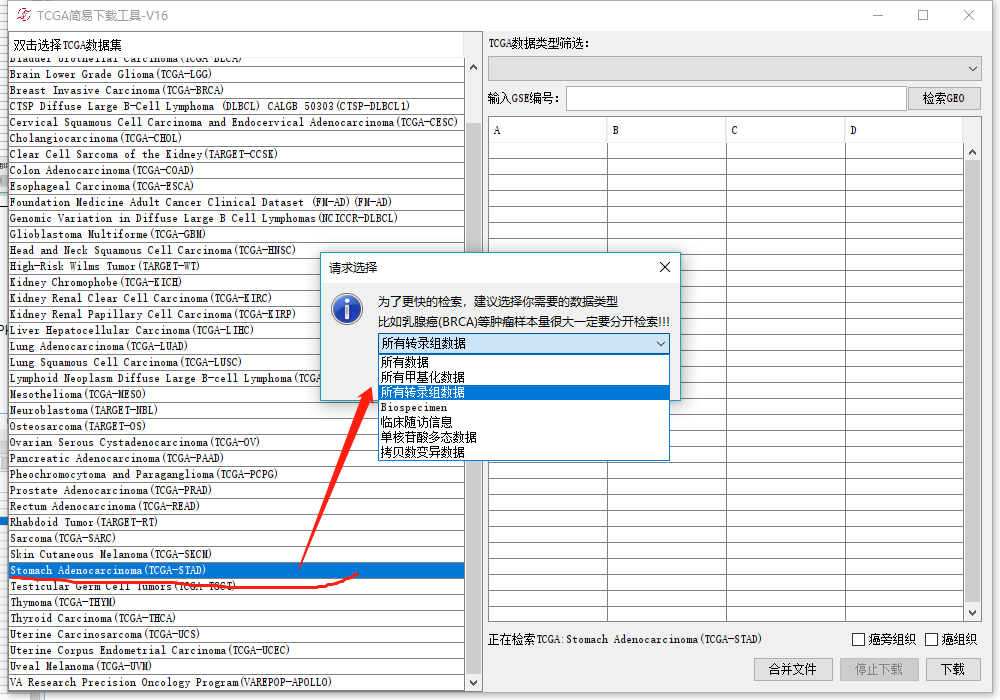 弹出一个小框,可以下拉,里面有多种选择,每一种选择都是检索一类数据,GDC做了限制,所以检索所有数据时如果样本量过大可能检索不出来,比如乳腺癌,所以这里分类型来检索比较实在。
弹出一个小框,可以下拉,里面有多种选择,每一种选择都是检索一类数据,GDC做了限制,所以检索所有数据时如果样本量过大可能检索不出来,比如乳腺癌,所以这里分类型来检索比较实在。
1、所有甲基化数据:主要包含两种平台的甲基化数据:27k和450k的
2、所有转录组数据:主要包含RNA-Seq,miRNA-Seq,划重点:下载基因表达、lncRNA表达、假基因表达、miRNA表达在这里下载
3、Biospecimen:主要是病人的入院收治信息
4、临床随访信息:主要包含病人的随访信息,划重点:你要的预后,生存,用药信息在这里
5、单核苷酸多态数据:主要包含SNP的数据,由四种软件处理的SNP数据都在这里面
6、拷贝数变异数据:主要包含CNV的数据,其中有两种一种是去除种系差异的,一种是没去除的。
我们先选择甲基化的看一看:
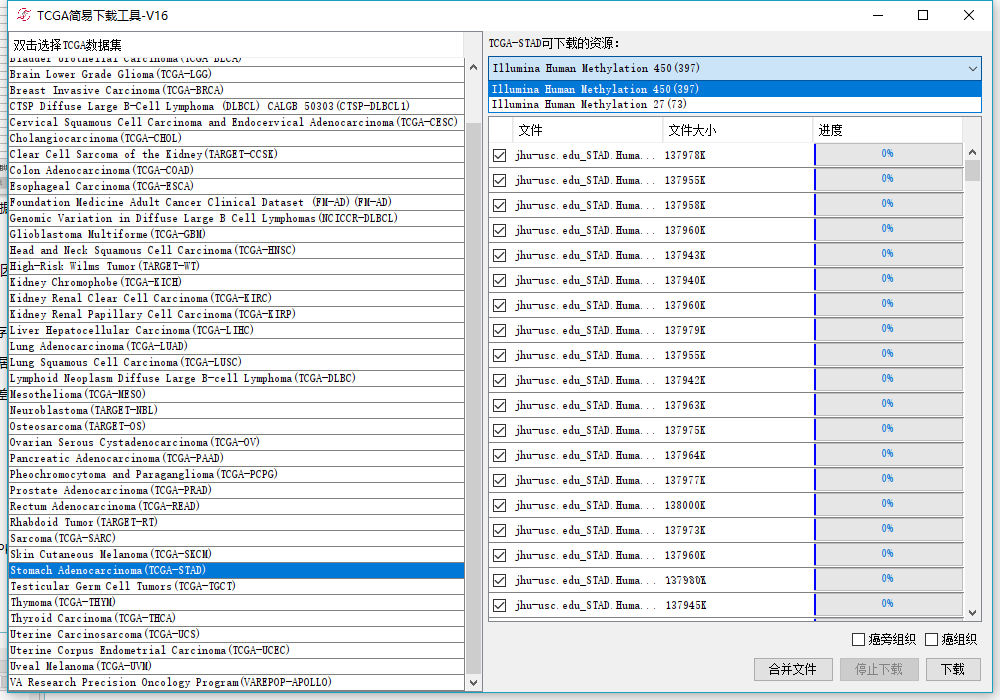 长这个样子,从中可以看到右上方下拉菜单有两种,450(397),27(73),这就说明胃癌 TCGA里测了两种平台的甲基化数据,样本量分别为397和73个,我们选择其中一个,然后右下角点击下载就好了,如果想进一步筛选一下癌症样本则右下角勾选癌症样本即可。
长这个样子,从中可以看到右上方下拉菜单有两种,450(397),27(73),这就说明胃癌 TCGA里测了两种平台的甲基化数据,样本量分别为397和73个,我们选择其中一个,然后右下角点击下载就好了,如果想进一步筛选一下癌症样本则右下角勾选癌症样本即可。
同理我们选择转录组数据看一看如下:
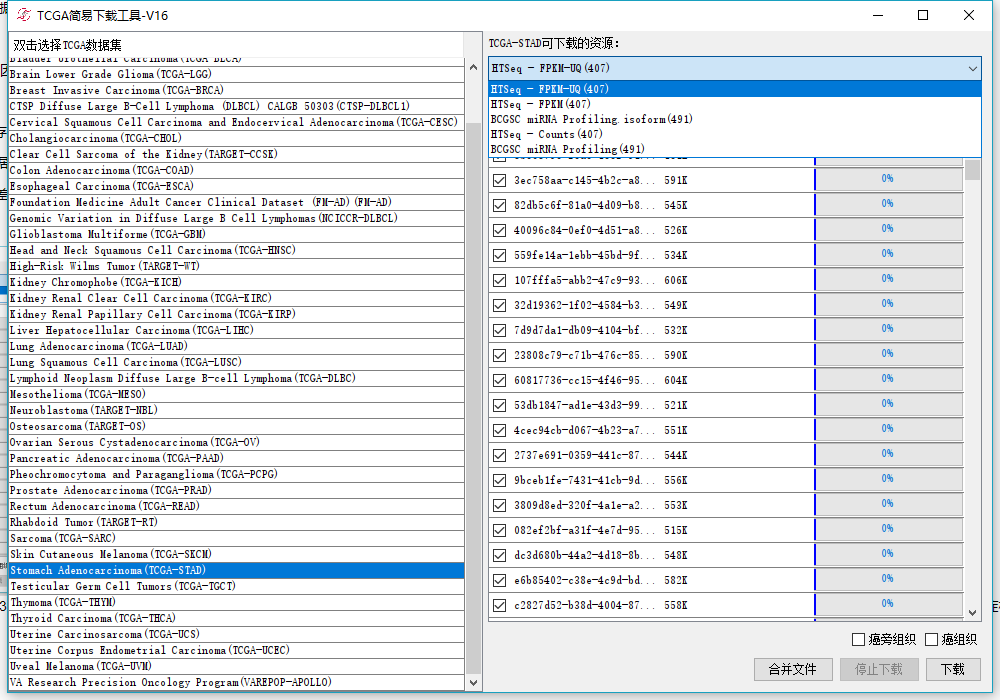 对!有五个,其中HTSeq 开头的表示的是RNA-Seq,三个分别表示三种定量方式,你酌情选择,不管选哪一种lncRNA、假基因、编码基因的表达谱都在这里面,BCGSC开头的表示miRNA-Seq,有两种,一种是isoform的这类是成熟体miRNA保存的地方,一种是不含isoform的,这类是前体miRNA保存的地方,你酌情选择,选择完右下角点击下载即可。
对!有五个,其中HTSeq 开头的表示的是RNA-Seq,三个分别表示三种定量方式,你酌情选择,不管选哪一种lncRNA、假基因、编码基因的表达谱都在这里面,BCGSC开头的表示miRNA-Seq,有两种,一种是isoform的这类是成熟体miRNA保存的地方,一种是不含isoform的,这类是前体miRNA保存的地方,你酌情选择,选择完右下角点击下载即可。
同理我们选择Biospecimen看看如下:
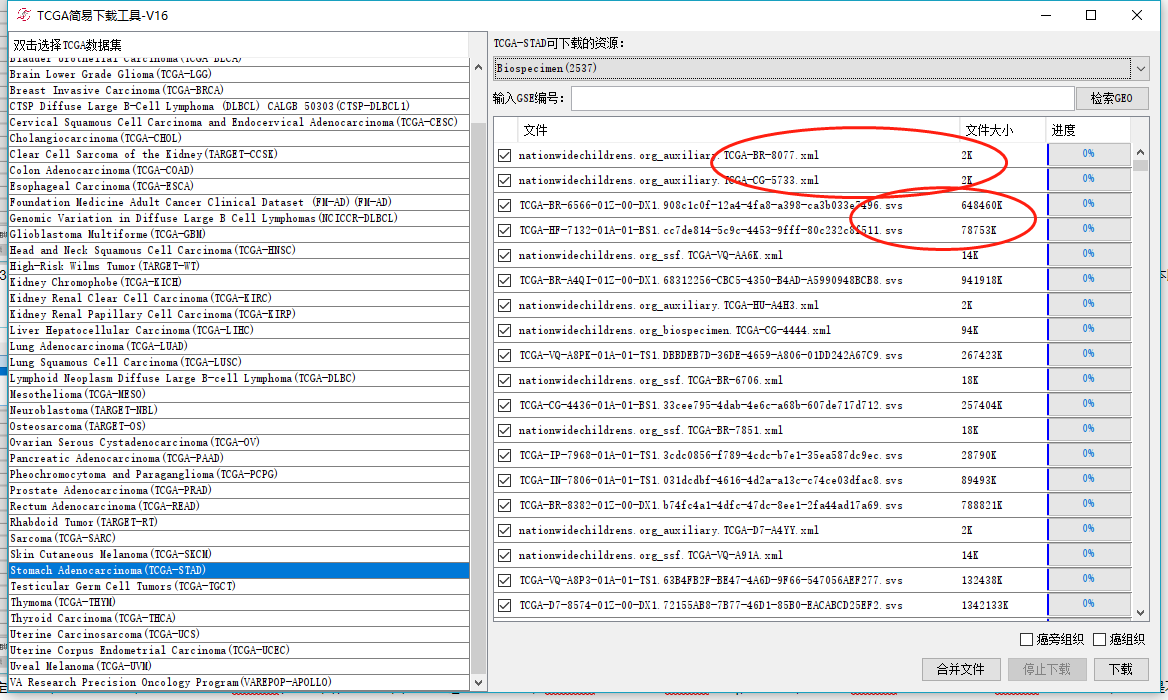 看一下,厉害了,有两千多个样本,这些都是啥呢,如图中红框文件大小比较小的,文件名是.xml结尾的此类是病人入院的信息,文件大小比较大的,文件名是.svs结尾的此类是病人的病理学图像数据,此类是最近新共享的,可以下载后用指定的软件打开看病理。
看一下,厉害了,有两千多个样本,这些都是啥呢,如图中红框文件大小比较小的,文件名是.xml结尾的此类是病人入院的信息,文件大小比较大的,文件名是.svs结尾的此类是病人的病理学图像数据,此类是最近新共享的,可以下载后用指定的软件打开看病理。
同理我们选择临床随访信息看一下:
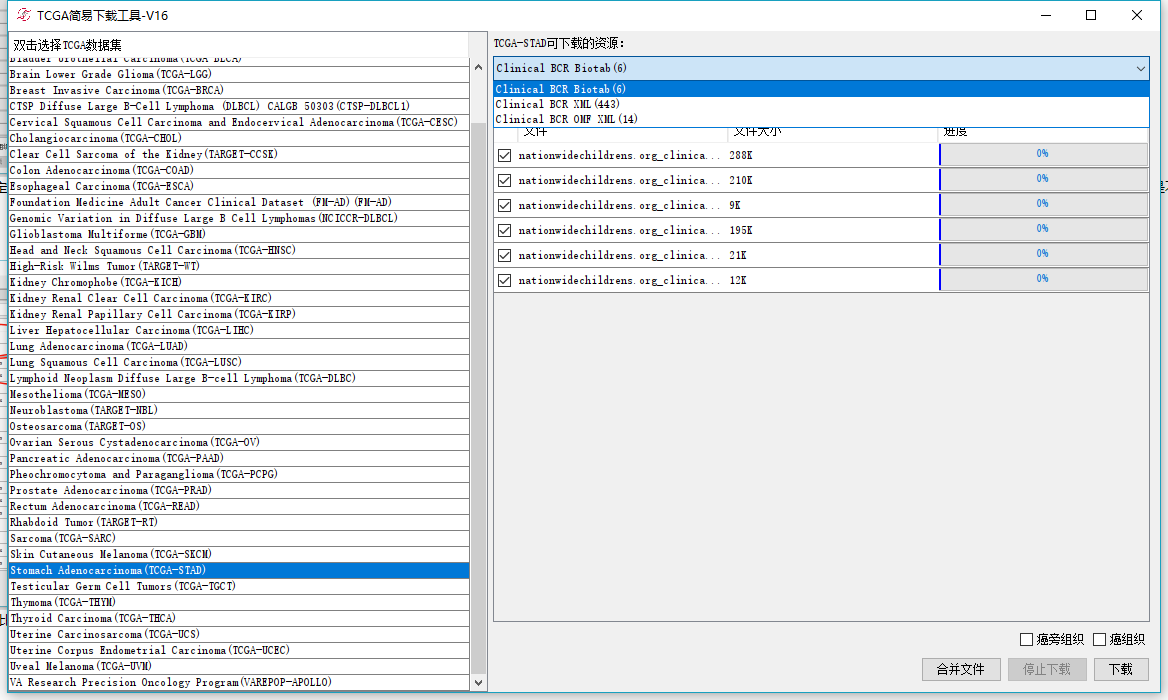
有三类,biotab,XML,OMF XML,第一个是病人的一些其他信息比如同时患了其他肿瘤的信息,是文本文件,而第二个才是随访信息,包含各种预后,治疗等信息的数据,第三个和第一个差不多,但是是xml格式的。
看一下单核苷酸多态的数据:
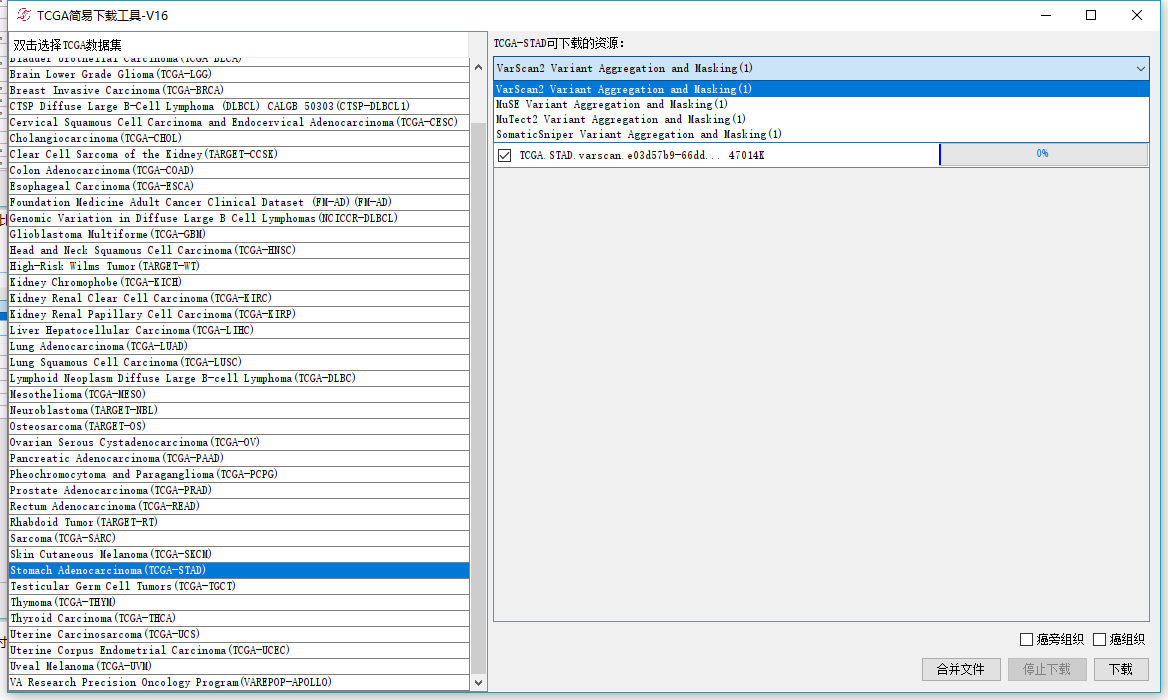 很明显是四个软件的结果,都是maf文件格式的,所有的突变都在一个文件里,要算TMP之类的可以在这里下载。
很明显是四个软件的结果,都是maf文件格式的,所有的突变都在一个文件里,要算TMP之类的可以在这里下载。
再看一下拷贝数变异数据:
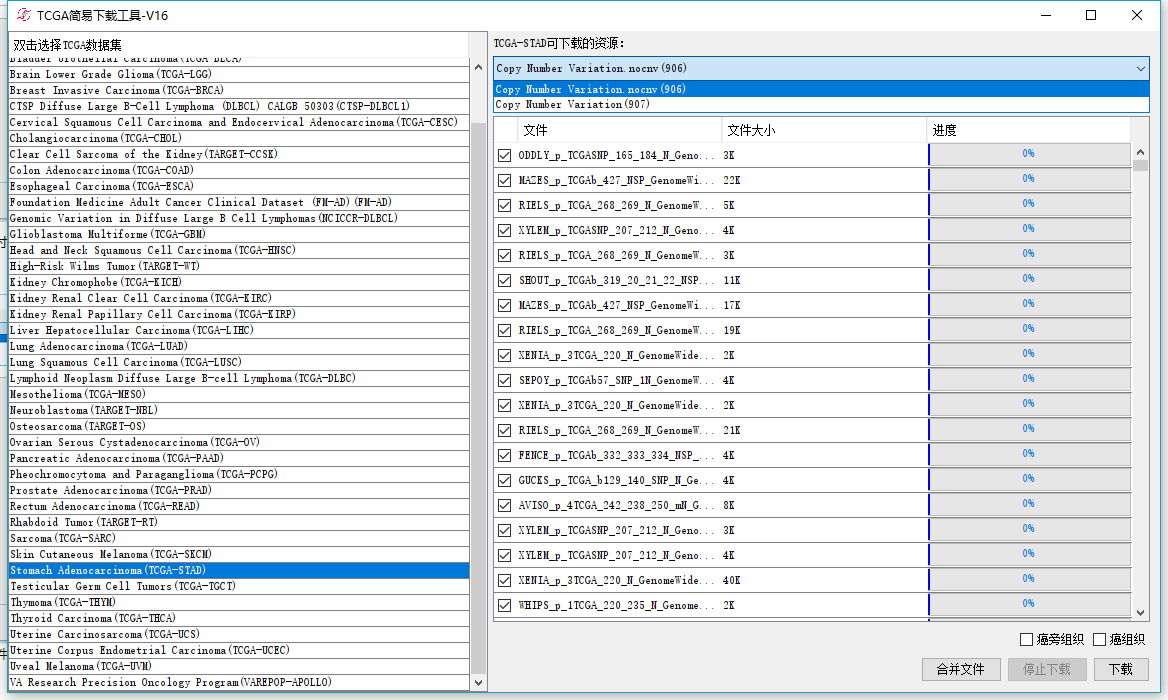 有两种,一种是.nocnv结尾的,一种是非nocnv的,样本个数是一样的,主要差别在于nocnv是去除了种系差异的,一般使用nocnv数据做后续的分析。
有两种,一种是.nocnv结尾的,一种是非nocnv的,样本个数是一样的,主要差别在于nocnv是去除了种系差异的,一般使用nocnv数据做后续的分析。
以上是所有类型的数据的下载介绍!数据下载完成之后大多数都不能直接用,故软件提供了 合并文件 这个功能,如右下角 合并文件按钮,以下载临床随访信息为例,数据下载完成是长这个样子:
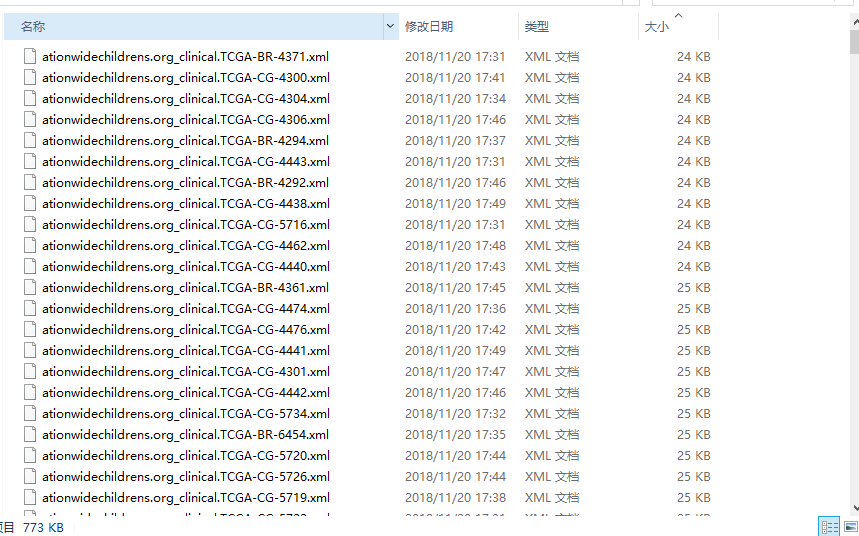
划重点:
除了单核苷酸多态、Biospecmen,其他各个类型的数据下载下来都是类似的 一个样本一个文件的样子,所以此时我们就需要将这些样本合并成矩阵,故点击“合并文件”按钮,弹出文件选择框,选择我们下载好的文件的文件夹,里面包含了一个.log结尾的文件,我们选择该log文件即可将这些样本进行合并,该log文件记录了本次下载中所有的样本的下载信息,所以软件根据该文件对这些样本进行合并成最终的矩阵。
与前几个版本的差别如:
一、miRNA数据合并时同时合并出两个矩阵,一个是counts的,一个是FPKM的
二、支持拷贝数变异的数据合并,可以用来直接跑GISTIC,或者做其他的下游分析
三、优化了临床随访信息中疾病进展状态(复发,转移等)及时间的提取,身高、体重、BMI等
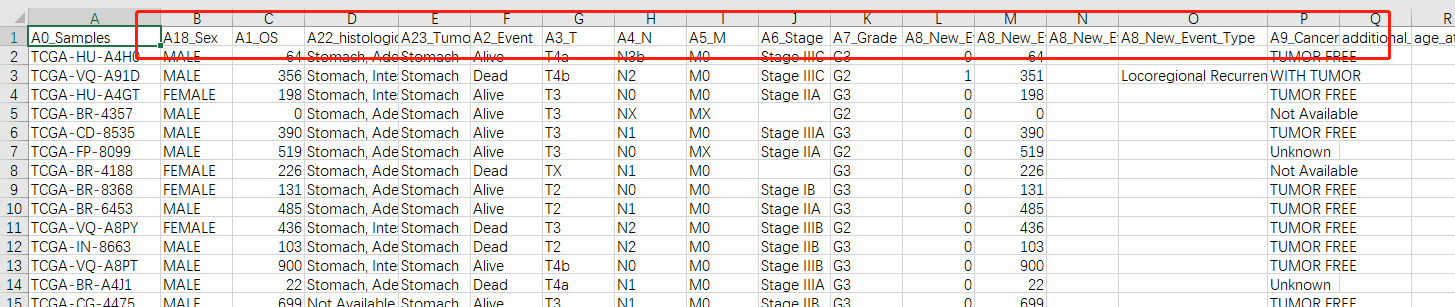 这些A开头的都是根据一定规则从随访信息数据中单独提取出来的,以便更轻松的往下分析。
这些A开头的都是根据一定规则从随访信息数据中单独提取出来的,以便更轻松的往下分析。
总生存时间和状态提取原则:死亡患者:首次出现死亡时的时间,未死亡患者:最后一次随访的时间。
进展时间和状态提取原则:进展患者:首次出现进展时的时间,未进展的患者:最后一次随访的时间。
- 发表于 2018-12-11 14:01
- 阅读 ( 13991 )
- 分类:软件工具
