这篇五分多的signature建模文章有点机智啊
最近偶然看到一篇生信建模的文章,思路清奇,让人茅塞顿开。

从标题上来看 是一篇分析胰腺癌甲基化异常的 基因的预后signature,这个题材的文章 看个标题就差不多能猜出来干了什么事,但好奇的我感觉发五分应该是有什么特别之处,于是仔细的看了一下,与大家分享。
本文整体思路简单锊了锊,大概如下:
1、下载ICGC数据库的甲基化数据和TCGA数据库的RNAseq数据
2、分别筛选差异甲基化位点和差异基因
3、紧接着 使用GEO的表达谱数据集GSE21501进行验证,验证这些种子基因的差异表达
4、将甲基化差异结果和基因表达差异结果关联,选择差异上调的基因并且启动子区域甲基化下调的基因和差异下调的基因并且启动子区域甲基化上调的基因作为候选基因(此步相似文章看这里:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5545832/)
5、对候选基因分别进行单因素生存分析(开始进一步找和预后相关的基因)
6、然后进一步使用lasso回归进行特征选择和构建预后模型
7、用验证数据集进行验证
8、分析模型在不同的临床分期中的预后预测效率
9、和已有的signature模型进行比较说明模型优势
10、功能富集分析模型中基因的功能
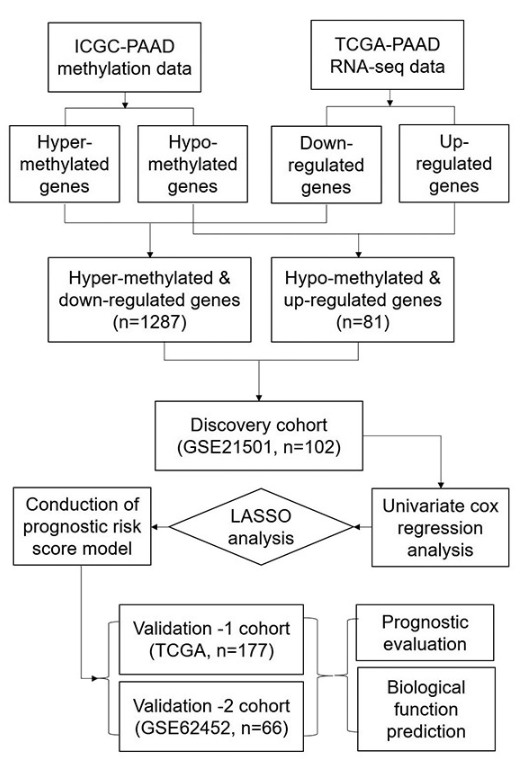
下面我们来看一下细节:
1、数据下载
从ICGC上下载DNA甲基化数据(https://www.garvan.org.au/research/cancer/pancreatic-cancer-research/)。
从TCGA上下载RNAseq数据(https://cancergenome.nih.gov/)及临床随访信息
从GEO上下载两个基因表达谱数据(GSE21501和GSE62452)(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21501,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62452)及对应的临床随访信息。
2、差异分析
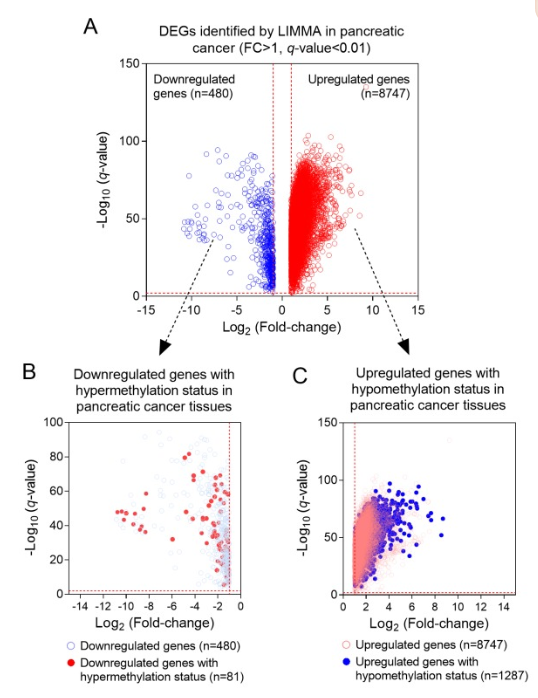
作者使用R软件包limma进行差异筛选,选择阈值FDR<0.01作为阈值,最终得到了9227 个差异表达的基因(图1A),进一步与ICGC的甲基化谱进行整合分析,确定了81个下调基因,这些基因是高甲基化的(81 / 480,16.9%)(图1B)和1287个低甲基化的上调基因(1287 / 847,44.7)(图1C)
疑问有三个:
1、方法学部分说用TCGA RNA-seq数据进行差异分析,利用GSE21501数据集进行验证,而这里结果只有GSE21501数据集的
2、FC>1 这个阈值是否太宽松了
3、甲基化的差异和基因表达差异用的样本不是同一个,准确来说:样本肤色都不一定一致,这里 推导出 受甲基化影响的基因表达 是不是有点远。
以上 真的可以这么来吗
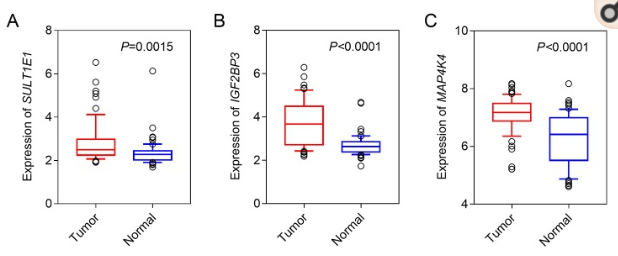
3、单因素生存分析
作者使用单因素cox分别对第二步中得到的1368个候选基因进行生存分析,选择p值小于0.05作为阈值,最终得到了三个基因:SULT1E1、IGF2BP3、MAP4K4。利用数据集GSE62452观察这三个基因在癌与癌旁中的表达差异
疑问:
1268个候选基因只有这三个基因的预后差异的吗?
为什么只得到了这三个基因呢
 4、使用lasso回归对这三个基因构建预后模型:risk score=0.195 * expression of SULT1E1+ 0.129 * expression of IGF2BP3 + 0.65 * expression of MAP4K4;并分别在验证数据集中验证验证。
4、使用lasso回归对这三个基因构建预后模型:risk score=0.195 * expression of SULT1E1+ 0.129 * expression of IGF2BP3 + 0.65 * expression of MAP4K4;并分别在验证数据集中验证验证。
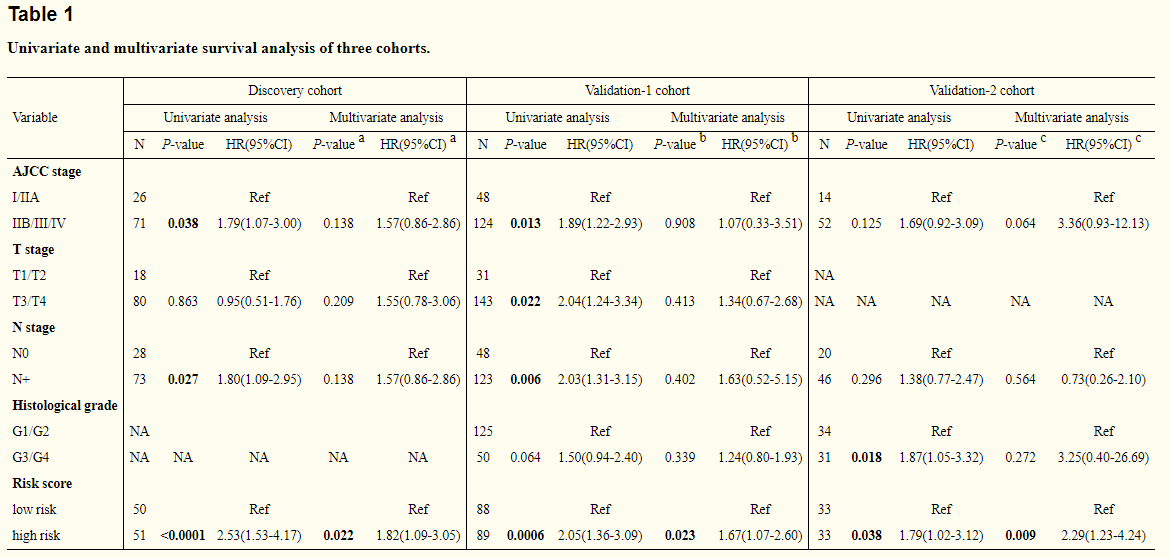
利用预后模型 分别将训练集和验证集数据代入模型计算样本的风险得分,再使用风险得分进行单因素和多因素生存分析,同时比较TNM分期在这三套数据集中的单因素和多因素生存分析结果,最终结果如Table1,从中可以看出模型在三套数据集中单因素和多因素都具有显著差异,同时与临床分期相比更显著
 作者进一步根据样本风险得分将样本平均分成高低两组,观察两组的预后差异,如图,从中可以看出三套数据集中也具有显著的预后差异
作者进一步根据样本风险得分将样本平均分成高低两组,观察两组的预后差异,如图,从中可以看出三套数据集中也具有显著的预后差异
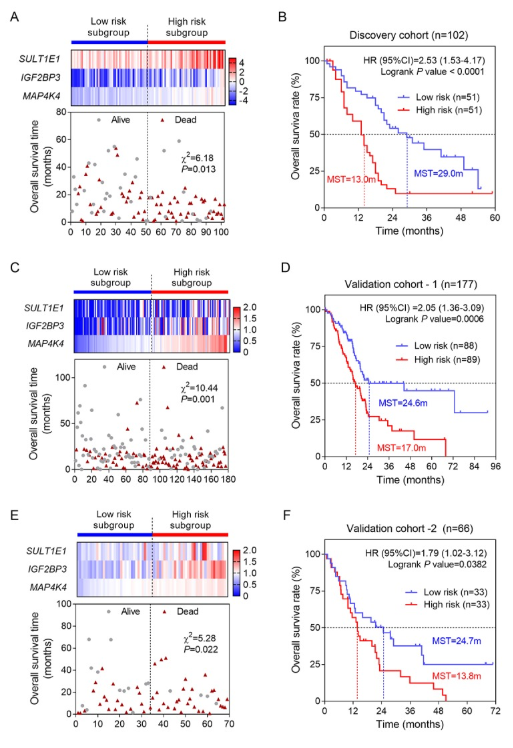 6、分层分析,观察模型在不同的临床分期中的预测效果
6、分层分析,观察模型在不同的临床分期中的预测效果
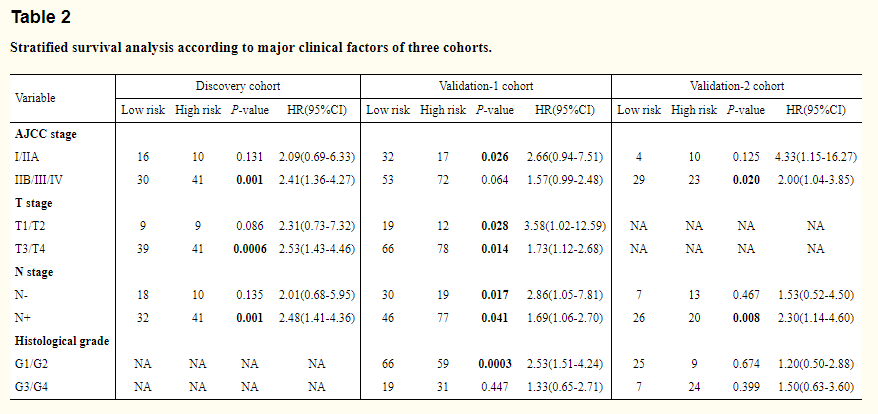
作者根据样本的临床TNM分期,分别将不同的分期样本单独拿出来使用模型去预后高低风险样本,结果如Table2所示,从中可以看出在多数临床分期中分类具有显著的差异,并且在T3/T4类型的样本中分类效果最好
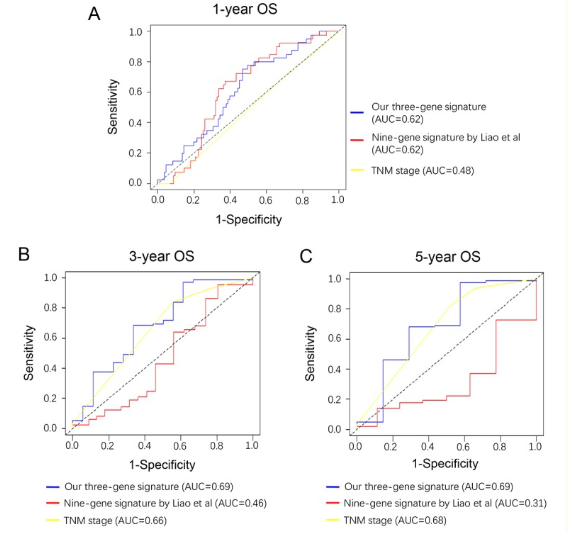
 7、与已知的模型进行比较
7、与已知的模型进行比较
作者选择了一个已经发表的利用TCGA的基因表达数据筛选的基因模型,分别比较了他们的1年、3年、5年的ROC,证明作者的模型线下面积最大,完胜他们。
疑问:
选一个两三分的文章的模型来比较来说明你的模型好,这是啥意思呢

8、功能富集
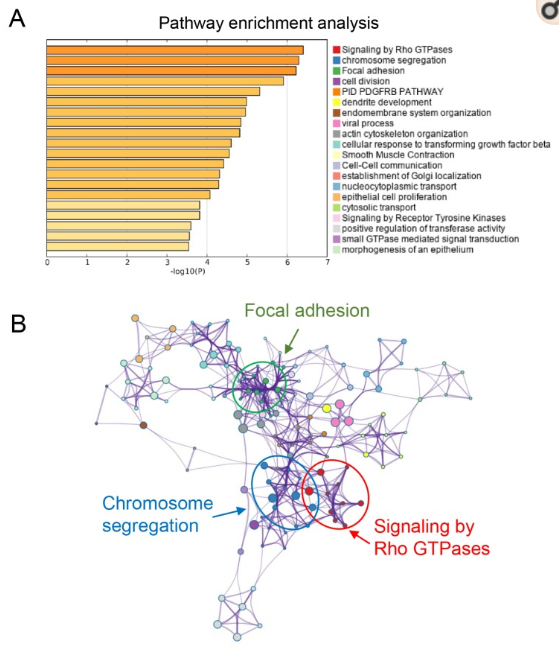
作者分别计算了这三个基因与基因组中其他基因的表达关系,选择最显著的前200个基因使用在线工具Metascape (http://metascape.org/)进行功能富集分析,发现最显著的前三个生物学途径是 Rho GTPases, chromosome segregation and focal adhesion pathways,已有报道表明这三个通路均参与肿瘤进展,为进一步研究胰腺癌的三基因模型的详细分子机制提供了依据。
以上便是文章的全部,总的来说 文章很多细节没有展示出来,有一种 欲盖弥彰的感觉,不过整个思路还是值得学习的。
- 发表于 2019-03-11 14:34
- 阅读 ( 11264 )
- 分类:方案研究
