那些年画过的热图之pheatmap美化过程
在基因的表达模式分析中,我们往往需要对多个基因表达数据进行可视化处理,使得我们所关注的基因在不同样本中表达情况一目了然。在日常研究中,我们往往习惯于选择热图实现这一基因表达模式可视化的需求,进而直观的表述我们的基因表达模式的分析结果。
推荐使用生信人的heatmap工具,更简单:http://gap.shengxin.ren/tool/3/
在基因的表达模式分析中,我们往往需要对多个基因表达数据进行可视化处理,使得我们所关注的基因在不同样本中表达情况一目了然。在日常研究中,我们往往习惯于选择热图实现这一基因表达模式可视化的需求,进而直观的表述我们的基因表达模式的分析结果。
今天就介绍一下非常简易使用的一个绘图包pheatmap
写在前面,准备一个表达谱矩阵,横轴为100个基因,纵轴为208个样本,如下:
 首先安装pheatmap
首先安装pheatmap
source("http://biocoundctor.org/biocLite.R")
biocLite("pheatmap")
然后加载
library(pheatmap)
最简单的调用如下:
pheatmap(profile)
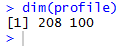
 长这个鬼样子简直没法看,从右侧可以看出数据跨度很大从0-200以上,图中大部分蓝色显示其实大部分值应该是在0-50这个区间,所以我们调整策略,取log(x+1)
长这个鬼样子简直没法看,从右侧可以看出数据跨度很大从0-200以上,图中大部分蓝色显示其实大部分值应该是在0-50这个区间,所以我们调整策略,取log(x+1)
pheatmap(log2(profile+1))
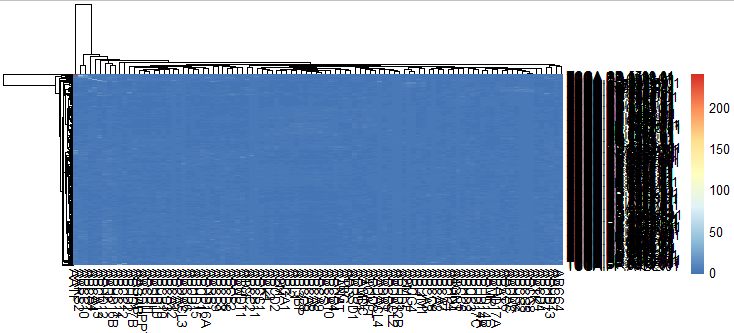
 比之前好看了点,但是还是有好多黄色,但是很明显看得出来聚类效果比之前好多了,但是很明显最小值太小以至于几乎看不到蓝色,颜色区分不开,进一步的来调整一下区间
比之前好看了点,但是还是有好多黄色,但是很明显看得出来聚类效果比之前好多了,但是很明显最小值太小以至于几乎看不到蓝色,颜色区分不开,进一步的来调整一下区间
bk = unique(c(seq(-5,5, length=100)))
pheatmap(log2(profile+1),breaks = bk)
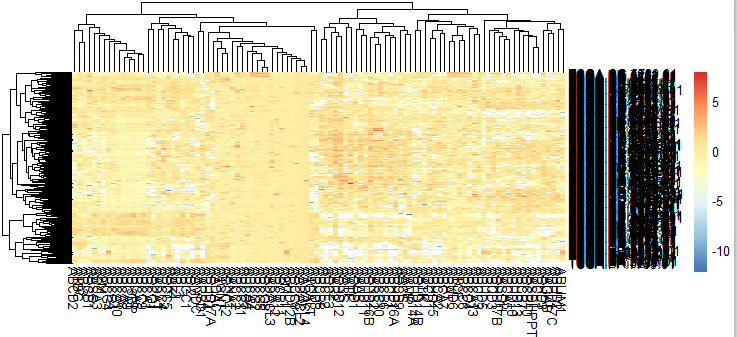
 此时看起来还不错,但是这个颜色有点不舒服,换个颜色试试
此时看起来还不错,但是这个颜色有点不舒服,换个颜色试试
bk = unique(c(seq(-5,5, length=100)))
pheatmap(log2(profile+1),breaks = bk
,color = colorRampPalette(c("navy", "white", "firebrick3"))(100))
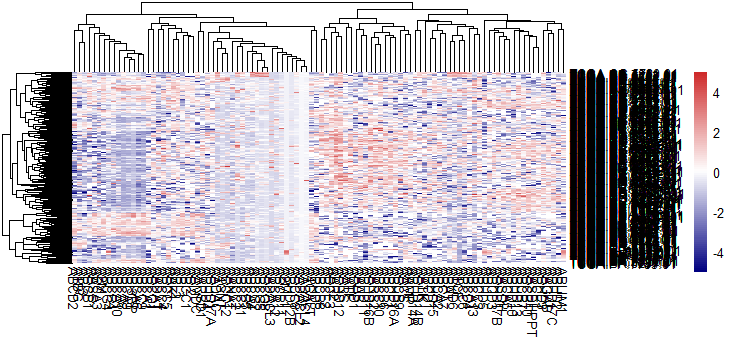
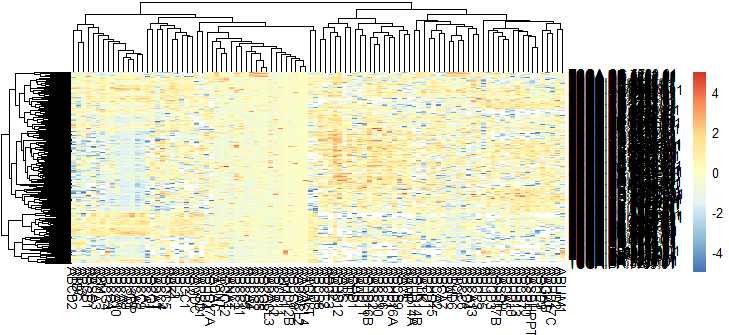
看起来还行,但是从图中可以看到那个中间那条一长条全是白色,事实上我想看看一个基因在不同样本中的高低,所以我应该使用纵轴的zscore进行标准化一下
bk = unique(c(seq(-5,5, length=100)))
pheatmap(log2(profile+1),breaks = bk,scale = 'column'
,color = colorRampPalette(c("navy", "white", "firebrick3"))(100))
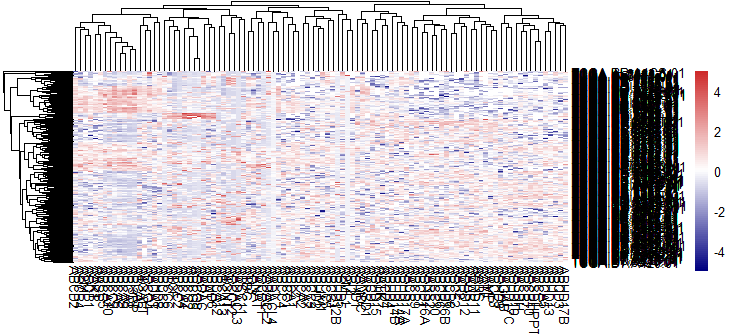 从图中可以看到有些基因在不同的样本中表达趋势比较一致的情况了,此时纵轴样本一团糊没什么意义,先把他去掉
从图中可以看到有些基因在不同的样本中表达趋势比较一致的情况了,此时纵轴样本一团糊没什么意义,先把他去掉bk = unique(c(seq(-5,5, length=100)))
pheatmap(log2(profile+1),breaks = bk,scale = 'column',show_rownames = F
,color = colorRampPalette(c("navy", "white", "firebrick3"))(100))

bk = unique(c(seq(-5,5, length=100)))
clust=pheatmap(log2(profile+1),breaks = bk,scale = 'column'
,show_rownames = F
,color = colorRampPalette(c("navy", "white", "firebrick3"))(100))
annotation_col = data.frame(
ClassGene = factor(paste0('Cluster',cutree(clust$tree_col,10)))
)
rownames(annotation_col) = colnames(profile)
pheatmap(log2(profile+1),breaks = bk,scale = 'column'
,show_rownames = F
,annotation_col = annotation_col
,color = colorRampPalette(c("navy", "white", "firebrick3"))(100))
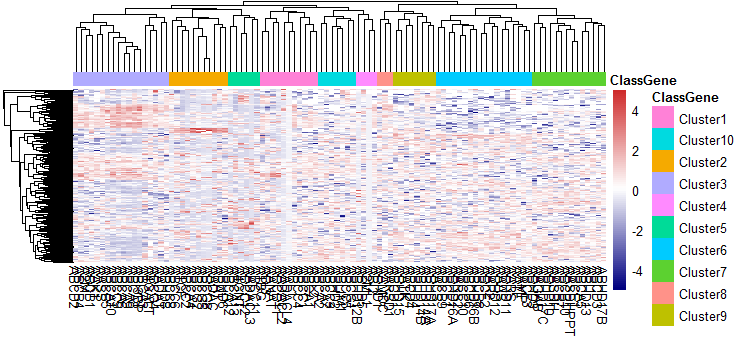
就写到这吧,还没有涉及的参数:
clustering_distance_rows = "correlation"#表示行聚类使用皮尔森相关系数聚类,当然也可以自定义如drows = dist(test, method = "minkowski");clustering_distance_rows = drows
cluster_row = FALSE#表示行不聚类
legend = FALSE#表示右侧图例不显示
display_numbers = TRUE#表示在热图中格子显示对应的数字,在那种横纵轴数目比较小是时候可用,比如样本间相关系数聚类
number_format = "\%.1e"#当显示数字时数字的显示方式
cellwidth = 15, cellheight = 12#表示热图中小方格的宽度和高度
fontsize = 8#表示热图中字体显示的大小
filename = "test.pdf"#表示直接就保存成test.pdf图片了
labels_row#可以自己定义横轴的显示字符,默认上图是基因名
main#类似title啦
gaps_col#产生一个间隔,就像有些文章中的那种分类后每个分类都有一个间隔
- 发表于 2017-06-26 20:57
- 阅读 ( 59759 )
- 分类:编程语言
