一篇曾经10+胃癌亚型预后相关的文章解析
今天给大家推荐一篇CCR上的文章(17年影响因子10+)

从标题来看做的胃癌亚型临场研究有意义,
n 研究背景:
胃癌在全球范围内最常见的癌症中排名第四,死亡率位居世界第三,每年约723,100人死于该疾病。2014年,TCGA数据库研究人员基于体细胞突变数据、mRNA表达谱数据、miRNA表达谱数据、拷贝数(CNV)改变数据以及蛋白质表达谱数据,采用决策树的方法将胃癌分成
i)Epstein–Barr virus (EBV)
ii)microsatellite instability (MSI)
iii)genomically stable (GS)
iv)chromosomal instability (CIN)这四种亚型[1]
但是在2014年的这篇文章中这四种亚型的临床特征并没有被进一步说明和研究,因此,来自德克萨斯大学安德森分校癌症中心的系统生物学部门的Bo Hwa Sohn等人,他们通过贝叶斯复合协变量的预测算法(Bayesian compound covariate predictor algorithms)对TCGA数据库中的262胃癌样本划分成上述的四种亚型(如Fig 1所示),并在样本数分别为267和432的两套独立数据集(MDACC和SMC)中去验证这些亚型的生存差异情况以及分析这些亚型是否会从辅助化疗中受益,最终构建了一个整合的胃癌风险评估模型。

Fig1.预测模型示意图
n 方法和材料:
(1) 数据的获取:从TCGA数据库下载262个胃癌样本的体细胞突变数据、mRNA表达谱数据、miRNA表达谱数据、拷贝数(CNV)改变数据以及蛋白质表达谱数据;将MDACC(GSE13861 & GSE26942; n = 267)和SMC(GSE26253; n = 432)数据集作为两个独立的验证数据集。
(2) 数据处理:对MDACC和SMC两套数据集进行四分位数标准化,再进行log2数值转化。
(3) 划分亚型:像这些文章所描绘的方法[1-9],基于多组学的数据(体细胞突变数据、mRNA表达谱数据、miRNA表达谱数据、拷贝数(CNV)改变数据以及蛋白质表达谱数据)对262个TCGA胃癌样本进行亚型的划分。其中,具有EBV特性的样本视为EBV亚型;具有高MSI的样本视为MSI亚型;通过体细胞突变和拷贝数改变数据将剩下的样本进一步划分为GS亚型和CIN亚型。
(4) 亚型特征基因的获取:基于各亚型的基因的mRNA表达谱数据,通过两两亚型之间做t.test检验,对于某一亚型来讲,只有该基因满足和其他三种亚型病人的表达中都差异的基因才认为是该亚型特异的基因(p < 0.001)。挑选各亚型中p值最显著的前200基因做后续分析(其中CIN亚型只有143个特异表达的基因,全部选取这143个基因)。
(5) 预测模型的构建:在262个TCGA胃癌样本中,基于各亚型特征基因分别去构建贝叶斯混合协变量预测模型(Bayesian compound covariate predictor algorithm),在各亚型预测模型中,以0.4为阈值划分该样本亚型,最终构建一个决策树预测模型。
(6) 验证数据集亚型划分:基于上述模型,对验证数据集进行亚型的划分。
(7) 生存分析:基于MDACC和SMC两套数据中各亚型病人的OS和RFS,对各亚型病人的生存进行log-rank检验。
(8) TCGA风险得分模型的评估:在上述TCGA以及MDACC和SMC两套数据中发现,EBV和MSI亚型病人的预后效果好,GS亚型病人的预后最差,CIN亚型病人和差的预后关系不是那么显著,因此构建风险得分模型如下所示:
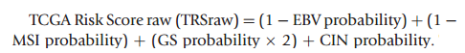
为了让该得分在0-100范围波动,最后的风险得分模型定义为:
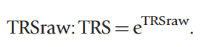
其中,low risk (<20), intermediate risk (20–30), and high risk of recurrence (>30)。
(9) 各亚型病人对辅助化疗药物的反应:比较恶性的病人更容易从辅助化疗药物中受益。在MDACC数据集中,先挑选AJCC stage为II、III或IV且没有转移的157个病人,在这157个病人中有116个病人接受了辅助化疗,这116个样本作为用药样本,映射到各亚型,得到各亚型中的用药组病人(CTX)以及非用药组病人(No CTX),基于log-rank检验各亚型中用药组病人与非用药组病人的生存是否差异。其中,CTX表示是接受了辅助化疗的病人组;No CTX表示没有采用辅助化疗的病人组。
n 结果展示:
(1)基于TCGA组学数据,最终将261个TCGA胃癌样本划分成四个亚型病人样本:EBV(n = 24),MSI (n = 57),GS(n = 54)和CIN (n = 127),其中,各亚型中特征基因表达结果如Fig 2所示。
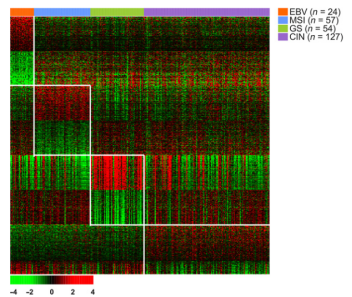
Fig 2. 各亚型特征基因表达情况
(2)各亚型预后评价:在两套验证数据集中,Bo Hwa Sohn等人发现EBV亚型病人预后最好,GC亚型病人的预后最差,MSI亚型和CIN亚型病人的预后比EBV亚型病人差,比CS亚型病人好(如Fig 3所示;在两套验证数据中的log-rank p value: p=0.004 & p=0.03)。
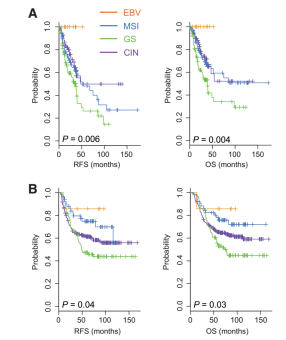
Fig 3. 两套验证数据集中各亚型生存曲线log-rank检验结果
(3)风险模型性能验证与评估:Bo Hwa Sohn等人又将MDACC和SMC两套验证数据集整合成一个大数据集(n = 699),想进一步去考量构建的风险得分模型评估预后的能力,他们将在TCGA训练数据集中得到的风险得分模型、T stage、N stage、AJCC stage、distant metastasis以及其他已知的胃癌预后因子作为协变量去构建多因素cox回归模型,在699个样本的验证集中去评估该风险得分模型,他们发现对TCGA样本所得到的风险得分可以作为独立的预后因素[HR = 1.5; 95% confidence interval (CI), 1.2–1.9; P = 0.001]。将该风险模型对n=699的验证集样本进行划分,将其划分为高、中以及低得分组病人,其中,得分小于20的划分为low组,得分介于20-30的划分为Int组,得分大于30的划分为high组,发现不同得分组别的病人在5年RFS和OS层面其log-rank p值显著(如Fig 4所示)。
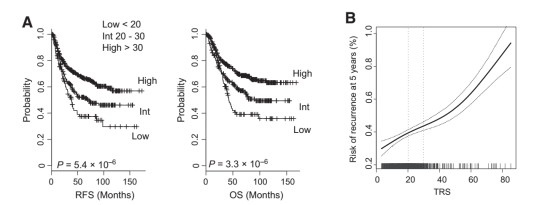 Fig 4. 风险预后模型log-rank检验.A)风险模型5年RFS和OS范围内,high,Int以及low-risk风险病人的差异;B)病人的5年复发率展示,其中两条虚线是95%置信区间。
Fig 4. 风险预后模型log-rank检验.A)风险模型5年RFS和OS范围内,high,Int以及low-risk风险病人的差异;B)病人的5年复发率展示,其中两条虚线是95%置信区间。
(4)辅助化疗药物对各亚型的影响:辅助化疗作为治疗胃癌的标准疗法,Bo Hwa Sohn等人又进一步刻画EBV、MSI、GS以及CIN亚型对辅助化疗的反应。在MDACC验证数据集中将各亚型中将样本分成CTX组和No CTX组,并通过log-rank检验两组病人生存差异(如Fig 5所示)。Bo Hwa Sohn等人发现,在CIN亚型病人中CTX和No CTX两组病人生存显著差异(p=0.03),这说明CIN亚型病人最能从辅助化疗药物中受益。
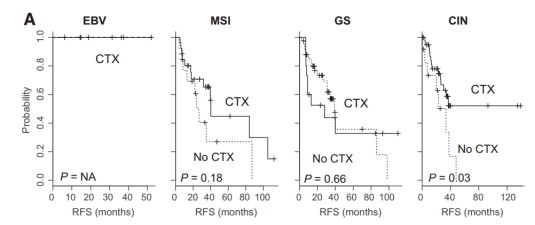 Fig 5. 各亚型中辅助化疗的影响
Fig 5. 各亚型中辅助化疗的影响
n 结论:
最终,通过分析TCGA的基因组数据和当前研究中产生的新数据,证明了4种胃癌亚型的临床意义,并开发了可以将胃癌患者可靠地分层为这4种亚型的预测模型。该预测模型不仅可用于识别预后不良的患者(GS亚型),还可用于识别最有益于辅助化疗(CIN亚型)的患者。
n 参考文献:
1. Network T C G A. Comprehensive molecular characterization of urothelial bladder carcinoma[J]. Nature, 2014, 507(7492):315-22.
2. Mclendon R , Friedman A , Bigner D , et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways[J]. Nature, 2008, 455(7216):1061-1068.
3. Bell D, Berchuck A, Birrer M J, et al. Integrated genomic analyses of ovarian carcinoma[J]. Nature, 2011, 474(7353): 609-615.
4. Koboldt D C, Fulton R S, Mclellan M D, et al. Comprehensive molecular portraits of human breast tumours.[J]. Nature, 2012, 490(7418): 61-70.
5. Muzny D M, Bainbridge M N, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer[J]. Nature, 2012, 487(7407): 330-337.
6. Hammerman P S, Voet D, Lawrence M S, et al. Comprehensive genomic characterization of squamous cell lung cancers[J]. Nature, 2012, 489(7417): 519-525.
7. Ley T J, Miller C A, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia[J]. The New England Journal of Medicine, 2013, 368(22): 2059-2074.
8. Getz G, Voet D, Lin P, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma[J]. Nature, 2013.
Weinstein J N, Akbani R, Broom B M, et al. Comprehensive molecular characterization of urothelial bladder carcinoma[J]. Nature, 2014, 507(7492): 315-322.
- 发表于 2019-06-28 10:17
- 阅读 ( 5231 )
- 分类:文献解读
