肿瘤TMB的计算原理及数学模型
作者按:继上周发表了“关于Omisc Core WES计算TMB的一点见解”(基因谷也同时转发了,在此表示感谢!),很多同行来联系并进行了深入交流。本人从事生物信息多年,最近花了近三年的时间夜以继日开发了一系列肿瘤NGS 检测算法(单计算函数自己就创建了近一百个!),包括超高深度测序数据的UMI分子标签算法。同时把这些算法集成到自主开发的云计算系统中。所有的计算都不需用复杂的命令行,简洁的界面化在线操作即可输出完美的报告结果。接下来,我会陆续将介绍计算肿瘤TMB的原理及数学模型,shell脚本计算流程,galaxy可视化编程以及自主开发的计算系统介绍给大家,并免费开放VarScanSomatic开源算法计算项目(http://www.genekras.com,仅代阿里云收取服务器费)给大家使用,可以进行Somatic SNPs and indel,CNV,MSI计算。
美国FDA目前批准了3个检测肿瘤TMB( tumor mutation burden)的方法:Omisc Core WES、MSK-IMPACT、FMI。如果按实验流程及操作复杂度来讲,这可能是有史以来最复杂的病理诊断方法。流程一般如下:手术取样、提取DNA、建测序文库、靶向捕获、NGS测序、数据比对、变异检测、变异注释、结果解读。复杂方法下的简单目的:数一数肿瘤特有的变异有几个,多数情况下平均每100万个位点还不超过10个变异。
TMB定义是:每Mb区域里非同义的体细胞变异个数。
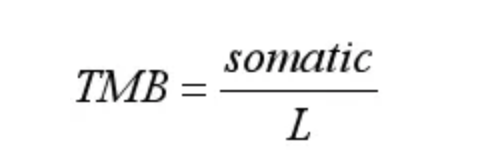
图1.somatic:检测到的非同义突变的体细胞变异个数。L:有效覆盖区域。
有效覆盖区域L容易知道,如果覆盖度足够好,可以用bed文件区间长度代替,那么计算TMB主要的工作就是怎么计算somatic。在肿瘤基因检测中计算somatic处于最核心的位置,靶向治疗、免疫治疗等都围绕着somatic展开。
Somatic定义为个体生长过程中产生的变异。目标就是要找在测序中肿瘤组织测到而正常组织没有测到的变异。因为测序有错误,如果测序足够深,肿瘤及正常组织大多数位点都会有变异。那么通过判断正常组织有无变异将不适合。我们可以模型化为肿瘤组织与正常组织某一变异有相同的频率为种系变异germline,否则为somatic。方法是对肿瘤组织及正常组织同时外显子靶向高通量测序,对每个位点进行比较。通过测序深度、频率等参数比较判断是否somatic。业内有很多计算软件可以用来计算somatic。下表是业内使用最多的开源软件[1],现在选取工业界使用最多的VarScan2 的算法来介绍somatic及其TMB的计算原理。
Variant caller | Statistical approach |
VarScan2 | Fisher's exact test |
Strelka | Bayesian algorithm |
Mutect2 | Bayesian algorithm |
表1 热门软件采用的数学模型
以下是VarScan2输出VCF格式文件位点的结果

SPV=8.8337E-2是用Fisher’s Exact Test计算的P-value值,用来判断是否somatic。那这值是怎么计算得到的,源文献没有具体说明,本人在这里给出计算过程。Fisher’s Exact Test接受2×2行列表作为计算对象,从上述结果提取数据可以得到下表。
tumor | normal | |
RD | 47 | 99 |
AD | 74 | 111 |
表2.肿瘤样本与正常样本覆盖深度2×2行列表。RD:支持参考序列的碱基数;AD:支持变异的碱基数。
Fisher’s Exact Test接受2×2行列表作为输入,并按如下方式计算P-value值:
1、计算行列表中每一行,每一列的总和以及观察总数。
2、给定行和列总和,如果原假设为真,则使用超几何概率函数计算条件概率,以观察行列表中的准确结果。条件概率为
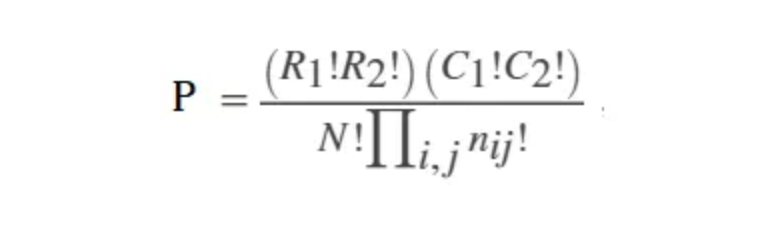
图2 条件概率计算公式
其中R1和R2是行总和,C1和C2是列总和,N是行列表中观测的总数,nij是表中第i行和第j列的值。
3、查找与行和列之和一致的所有可能的非负整数矩阵。对于每个矩阵,使用P公式计算相关的条件概率。
4、根据感兴趣的替代假设,使用这些值来计算检验的p值。
(1)对于双边测试,对于观察到的行列表,将所有小于或等于Pcutoff的条件概率求和。
(2)对于左侧测试,将(1,1)象限频率小于或等于n11的所有矩阵的条件概率求和。
(3)对于右侧测试,在观察到的行列表中求和(1,1)单元频率大于或等于n11的所有矩阵的条件概率。
VarScan2 somaitc计算的是单侧测试,如果按下表排列数据是左侧测试。
tumor | normal | |
RD | 47 | 99 |
AD | 74 | 111 |
表2.肿瘤样本与正常样本覆盖深度2×2行列表。
那怎么计算P-value呢?根据P-value的定义:在原假设为真的前提下,出现该样本或比该样本更极端的结果的概率之和。上表就是所有tumor Ref depth小于等于47的概率之和。

(P根据公式1计算)
P-value=0.0883,这P的意思是假设tumor Alt与normal Alt有同样的频率,但这的假设可能性为0.0883。按通常0.05或0.01阈值来cutoff的话,应该接受这一假设。肿瘤样本中测到的位点是种系变异而非somatic。
再看一个测序位点

tumor | normal | |
RD | 144 | 230 |
AD | 39 | 0 |
表3.肿瘤样本与正常样本覆盖深度2×2行列表。
P-value=1.3226e-15,这P值就足够小,认为是种系变异就不合适了,可以判断为肿瘤特有的变异somatic。
按照上述方法对肿瘤样本与正常样本所有同时覆盖的位点都计算一个P-value值,当P-value值小于设定值时判定位肿瘤体细胞变异。因影响NGS测序结果的因素多,reads覆盖的概率分布并不跟理想模型一致。如果按照常见0.05值来判定somatic,结果会比预期多。可以通过更低的P-value值或其他条件过滤掉可能的假阳性位点。按本人计算经验,P-value设0.001跟mutect2设的默认参数差不多。如果somatic是10个,覆盖到的区域2M,那么TMB=10/2=5。(敬请期待下期:TMB计算流程实现。)
参考资料:
Anne Bruun Krøigård Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS One. 2016; 11(3): e0151664.
Daniel C. Koboldt.VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing.Genome Res. 2012 Mar; 22(3): 568–576.
作者:刘远东:广州克拉斯基因科技有限公司创始人,算法研发工程师,有多年生信算法开发经验。广州克拉斯基因科技有限公司专注肿瘤云计算,国内首家提供UMI标签测序数据在线计算的系统平台!扫描下方二维码可随时与作者全方面交流。零成本生信计算助您成功走在同行前列!点击进行计算http://www.genekras.com。
如果你对本系列有兴趣可以考虑加群:

也可以添加小助手拉你进群:

- 发表于 2019-12-09 11:07
- 阅读 ( 8150 )
- 分类:基因组学
