如何解决生信新手分析时常常遇到的数据问题
点击"生信学霸"添加关注,我们一起进步
“高通量测序后续分析常见的数据格式”
前段时间我们推送了很多小工具教程;
点击上方“SangerBox工具使用讲解”获取更多工具讲解
从小伙伴们使用情况和私底下小伙伴反馈过来的情况来看,主要的问题是很多小伙伴刚刚入门生物信息,对现阶段分析所用到的数据格式不是很清楚。
今天小编就和大家聊一聊分析时常用的数据格式,干货满满,建议收藏与转发。
—
生信分析
其实生信分析分为标准分析、高级分析和个性化分析。
所谓的标准分析就是从最原始的fastq文件进过质控,过滤到低质量的数据,在mapping到人类参考基因组上,最后进行数据定量,转换得到表达矩阵的过程。
这个过程的技术特点由于测序原始数据比较大,单个文件往往在10G左右,大多数由服务器上完成,与高级分析、个性化分析也相比,所涉及到的软件和参数比较固定,对计算资源要求很高,部分数据对应TCGA上level1、level2的数据。对于我们生信初学者拿到的数据是表达定量之后的表达谱数据,可以进行高级分析和个性化分析,这两部分对分析者的技能和知识水平较高,同时分分析手段和方法也比较多,可以选择R语言、python、在线云平台以及图形化软件,就是所谓的“挖掘机技术哪家强”。
—
基础概念
Reads:字面可以翻译成“读段”,测序仪上测出的一小段DNA序列,是测序的最小单位。
Fragments:由于二代测序读长较短,例如:Illumina测序仪单段、端长度只有100bp或150bp,所以在测序之前将DNA或RNA打成小的片段,这些小的DNA片段就是fragments。测序结果可分为单端测序和双端测序,对于单端测序只能从fragments的一端测序,测出的reads数目与fragments数目相等;对于双端测序会测到fragments的两端,会得出两个reads,所以reads的数据是fragments数目的两倍。
测序深度:是指测序得到的碱基总量(bp)与物种基因组大小的比值,或者理解为基因组中每个碱基被测序到的频数。
测序覆盖度:是指测序得到的序列占整个基因组的比例。或者可以理解为基因组被测序到的碱基个数,占整个基因组的比例。
—
常见的数据格式
1. Count数据
----------
定义:高通量测序中比对到参考基因组中exon上的reads数,得到的基因表达矩阵是简单计数得来的,因此称作原始计数,也称Count矩阵。可以用作差异分析,常用的软件有edgeR和DEseq2
优点:能有效说明某个区域是否真的有表达和真实的表达丰度,能够近似呈现真实的表达情况,有利于实验验证。
缺点:因为exon长度不同,不同exon无法进行丰度比较;由于测序总数不同,难以对不同测序样本间进行比较。
2. FPKM/RPKM
----------
由于上面用count数据作为某个转录本的丰度,由于不同的exon长度不一样被测序到的次数也不一样,一般来说,exon的长度越长,产生的reads数据也就越多,相应的丰度也越高,不同样本上机批次不同,测序深度也会有所差异,所以需要将不同长度exon和测序深度不同的reads进行标准化,使数据的表达值在同一“起跑线”上。所以同行研究者们定义了FPKM/RPKM对数据进行标准化。
RPKM: Reads Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的reads),主要用来对单端测序(single-end RNA-seq)进行定量的方法;
total exon reads:某个样本mapping到特定基因的外显子上的所有的reads
mapped reads (Millions) :某个样本的所有reads总和
exon length(KB):某个基因的长度(外显子的长度的总和,以KB为单位)
FPKM: Fragments Per Kilobase of exon model per Million mapped fragments(每千个碱基的转录每百万映射读取的fragments),主要用来对双端测序(par-end RNA-seq)进行定量的方法;[1]。
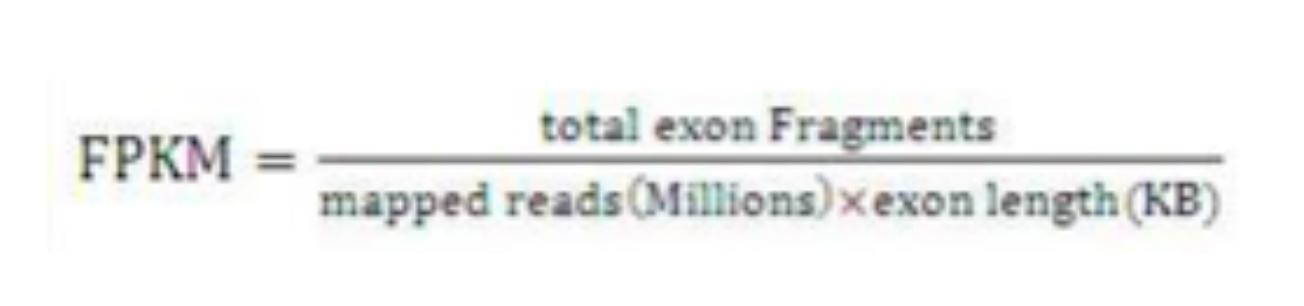
FPKM和RPKM的原理是相似的,两者的区别在于,对于单末端测序数据,由于Cufflinks计算的时候是将一个read当做一个fragment来算的,故而FPKM等同于RPKM。对于双末端测序而言,如果一对paired-read都比对上了,那么这一对paired-read称之为一个fragment,而如果一对paired-Read中只有一个比对上了,另外一个没有比对上,那么就将这个比对上的read称之为一个fragment。而计算RPKM时,如果一对paired-read都比对上了会当成两个read计算,而如果一对paired-read中只有一个比对上了,另外一个没有比对上,那么就计read数为1。故而即使是理论上将各个参数都设置成一样的,也并不能说FPKM=2RPKM。
3. TPM
----------
定义:Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)
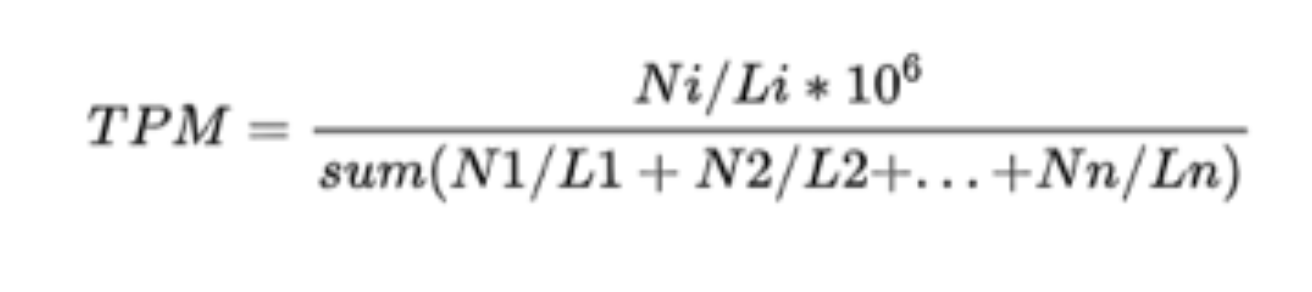
Ni:比对到第i个exon的reads数
Li:第i个exon的长度
sum(N1/L1+N2/L2 + ... + Nn/Ln):所有 (n个)exon按长度进行标准化之后数值的和
4. 总结
----------
TPM、RPKM、FPKM三者的区别是计算次序不一样。TPM可以看作是RPKM/FPKM值的百分比。当计算TPM的时候,先对基因长度进行归一化,其次是测序深度的归一化。当使用TPM时候,每个样本的TPM总和是一样的。这使得比较同一个基因的reads数在不同样本间的比例变得容易。TPM实际上改进了RPKM/FPKM方法在跨样品间定量的不准确性。FPKM和RPKM与此相反,每个样本的FPKM或RPKM的累加和可以不一样,造成样本间不能直接比较FPKM或RPKM值。
参考文献
Wagner, G.P., Kin, K. & Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
具体指引详见:

- 发表于 2020-06-15 15:03
- 阅读 ( 7687 )
- 分类:软件工具
