What?PC跑转录组。
What?PC跑转录组。
简介
上次小编介绍了如何利用生信人小工具进行芯片数据的处理。但是相信大家有些时候还是需要从转录组数据开始的。
应用场景:比如之前的数据大家只是对编码的基因进行注释,非编码RNA(lncRNA)并没有处理,这是巨大的浪费;还有就是比如人的基因组,现在又更新了好几个版本,有一些新的基因出来了,当你需要对新基因研究时,你可能需要重新做定量;最最重要的就是当你发现一个特定条件下物种,或者一个特定处理的样品,就只有一套转录组数据,在这非常坑爹的时候,是必须必须必须要从头进行转录组定量的。
然后,芯片数据处理可以在个人笔记本上跑,芯片数据可以吗。
当然可以了。
知识点
给大家介绍干货之前,先给大家推荐另一个干货。
一篇新的文章,Alignment-free sequence comparison: benefits, applications, and tools
这篇文章小编已经拜读过了,讲得是两种比对算法的区别,一种是base-alignment,另一种是alignment-free,而且作者还力推alignment-free会在应用端有很好的发展。
Kallisto介绍
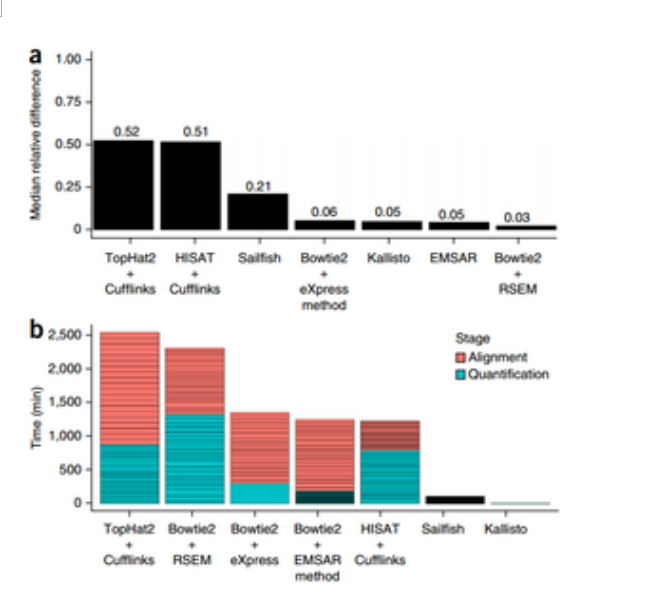
说完这些,跟大家介绍下今天的主角kallisto,该软件是基于alignment-free思想的有参转录组定量软件,号称10分钟内完成30Mb Reads的序列定量。
传统的比对是将reads分割成k-mer后,将每一个k-mer分配到hash表中一个唯一的位置,再进行序列比对。通过这种转换,可以大大提高序列比对的效率。当存在k-mers可以比对到基因组的不同位置上的情况时,就会降低定量分析的准确度。但是Kallisto有效地解决了这个问题。Kallisto并不需要知道Reads来源于转录本的具体位置,只要知道是哪个转录本就可以精确定量(着重于确定一个 read 属于哪一个基因,而不关心 read 在基因上的位置)。
界面版的Kallisto
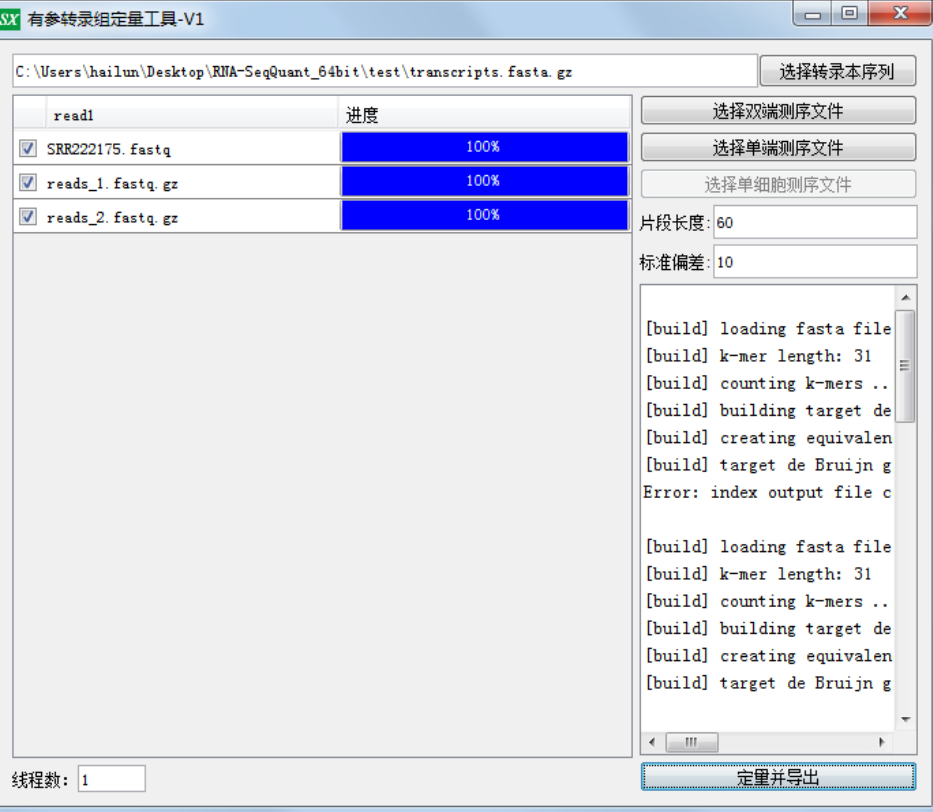
然后该软件有windows版的,当然了小编考虑到各位小白的入门问题,就随手开发成了界面版的。
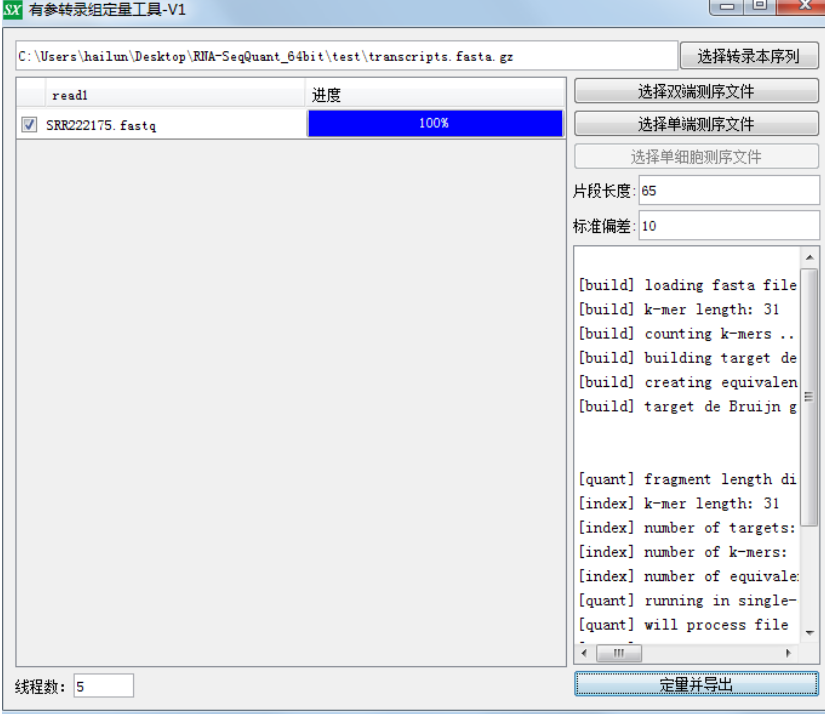
软件使用很简单,首先是选择转录本序列,这个可以去ensemble或者ncbi去下载,然后要注意自己的fastq 是单端的还是双端的,如果是双端的话,不需要处理,但是如果是单端的需要填写建库大小和标准偏差。
亲测有效
另外,小编亲测,read数据量为7.9Gb,跑人的转录本定量(5万多个),最高内存没有超过4Gb。
所以说大家就妥妥的用吧。
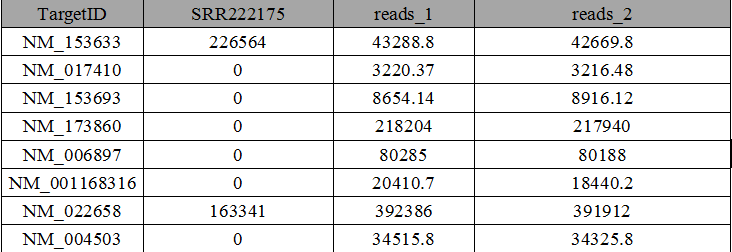
还可以一次性跑多套数据哦(方法是直接选中多套fastq数据即可),直接生成表达矩阵。
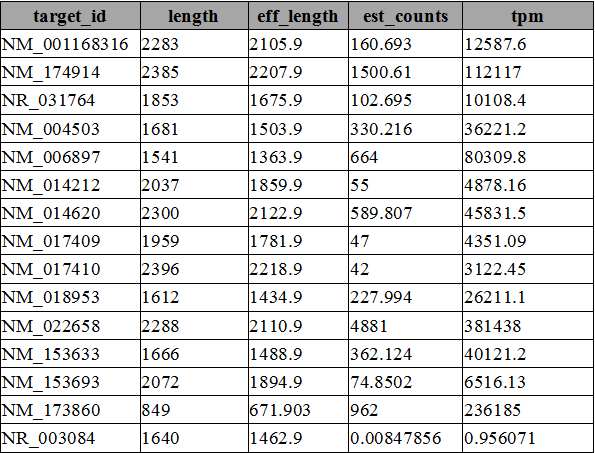
结果:
你可能还需要
另外,如果你可能需要去下载sra数据。
首先打开NCBI 去下载sra数据压缩包,例如
ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP118/SRP118996/SRR6107775/SRR6107775.sra
然后去下载 sratoolkit,这个也是免安装的。
然后去CMD下面运行fastq-dump *sra 将sra转化为fq数据。(这一步马上开发成界面,请各位稍等)
然后拿到fastq数据之后,就可以直接利用上面的软件去做定量了。
- 发表于 2017-10-23 17:09
- 阅读 ( 7693 )
- 分类:软件工具