关于TCGA RNA-Seq数据ID的转换方法
使用TCGA简易下载工具的同志们一直很好奇,RNA-Seq数据集中的ID是怎么转换的,今天就介绍一下原理
首先我们根据TCGA官网提供的RNA-Seq pipline(https://docs.gdc.cancer.gov/Data/Bioinformat...
使用TCGA简易下载工具的同志们一直很好奇,RNA-Seq数据集中的ID是怎么转换的,今天就介绍一下原理
首先我们根据TCGA官网提供的RNA-Seq pipline(https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/)里面可以看出TCGA使用的gtf是从gencode上面下载的,如图:
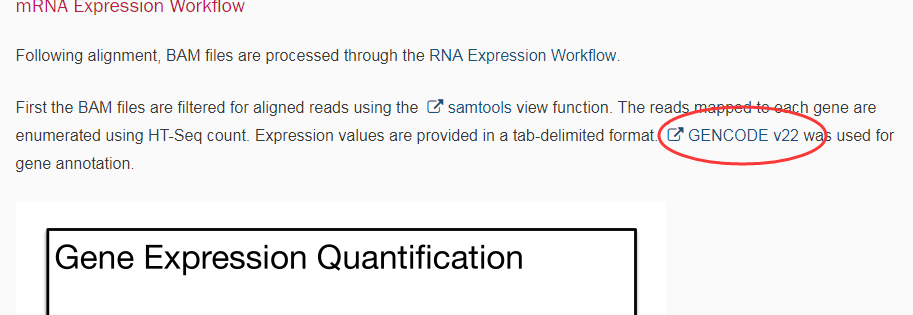
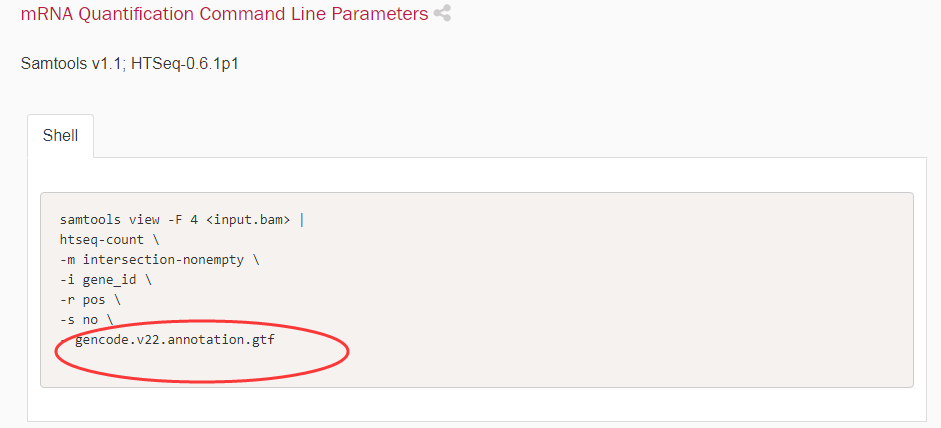 所以我们去GENCODE(http://www.gencodegenes.org/releases/22.html)上下载了对应的gtf文件,如图
所以我们去GENCODE(http://www.gencodegenes.org/releases/22.html)上下载了对应的gtf文件,如图
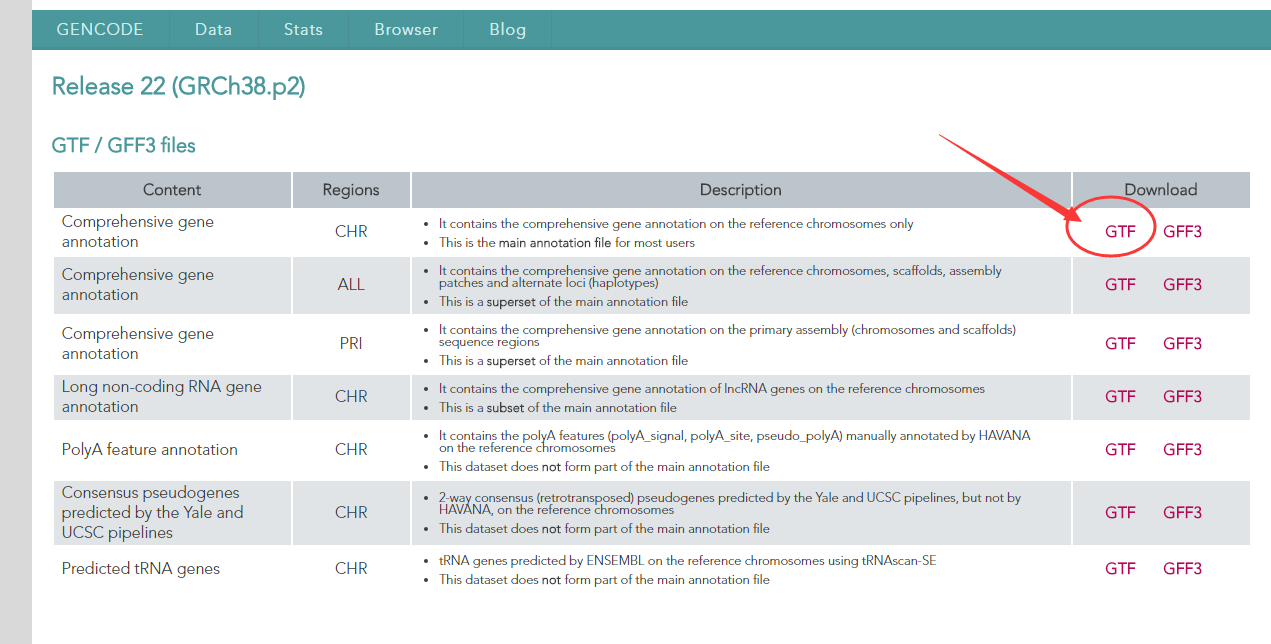 下载完之后是一个标准的gtf格式的数据,我们进一步解析这个数据,写一个简单的统计小脚本统计一下里面的基因个数
下载完之后是一个标准的gtf格式的数据,我们进一步解析这个数据,写一个简单的统计小脚本统计一下里面的基因个数
f=open('gencode.v22.annotation.gtf')
line=f.readline()
_list=[]
n=0
while line:
line=line.strip()
if line.find('#')!=0:
cols=line.split('\t')
_list.append(cols[2])
if cols[2]=='gene':
#print cols
n+=1
line=f.readline()
f.close()
print(set(_list))
print n
 可以看出里面的基因个数是60483,与TCGA里面下载的RNA-Seq中的ENSG编号个数一模一样
可以看出里面的基因个数是60483,与TCGA里面下载的RNA-Seq中的ENSG编号个数一模一样
打开里面的文件可以看到有基因的描述文件如下:

其中包含:
1、gene_id=ENSG 也就是TCGA RNA-Seq中使用的ID
2、gene_type 也就是用来描述这个基因的属性,一般为编码基因,miRNA,lncRNA,假基因等,我们可以根据这个信息提取lncRNA的表达谱
3、gene_name 也就是大家常见的gene_symbol
使用以上信息就可以进行TCGA的ID转换了
至于lncRNA的定义可以参考这里:https://www.shengxin.ren/question/23
最终提取各个属性的ID对应关系的代码如:
f=open('gencode.v22.annotation.gtf')
line=f.readline()
_list=[]
fw=open('GeneTag.txt','w')
fw.write('ENSGID\tSYMBOL\tTYPE\tLoc\tGENESTATUS\tGENETYPE\n')
while line:
line=line.strip()
if line.find('#')!=0:
cols=line.split('\t')
_list=[]
if cols[2]=='gene':
_ids=cols[8].split(';')
ensg=_ids[0][_ids[0].find('ENSG'):_ids[0].rfind('.')]
gene_type=_ids[1][_ids[1].find('"')+1:_ids[1].rfind('"')]
gene_status=_ids[2][_ids[2].find('"')+1:_ids[2].rfind('"')]
gene_name=_ids[3][_ids[3].find('"')+1:_ids[3].rfind('"')]
_list.append(ensg)
if gene_status=='NOVEL':
_list.append(ensg)
else:
_list.append(gene_name)
if gene_type=='processed_pseudogene' or gene_type=='unprocessed_pseudogene' or gene_type=='pseudogene' or gene_type=='unitary_pseudogene' or gene_type=='polymorphic_pseudogene':
_list.append('pseudogene')
elif gene_type=='lincRNA' or gene_type=='sense_intronic' or gene_type=='sense_overlapping' or gene_type=='antisense' or gene_type=='processed_transcript' or gene_type=='3prime_overlapping_ncrna':
_list.append('lncRNA')
elif gene_type=='protein_coding':
_list.append('protein_coding')
else:
_list.append('Other')
_list.append(cols[0]+":"+cols[3]+"-"+cols[4]+cols[6])
_list.append(gene_status)
_list.append(gene_type)
fw.write('\t'.join(_list)+'\n')
line=f.readline()
f.close()
fw.close()
引用:转录本属性定义http://vega.archive.ensembl.org/info/about/gene_and_transcript_types.html
- 发表于 2018-01-14 19:18
- 阅读 ( 19413 )
- 分类:方案研究
