干货:如何将别人的芯片数据唯我所用,发自己的文章
人类基因组目前已经更新到GRCh38,相较于之前版本必然是有它优化的地方,但是在long long ago的芯片数据在进行注释时必然使用的不是最新版本的基因组,那么如何将这些芯片数据与门当户对(相匹配),请您继续往下观看
目的:就是将原本为mRNA设计的芯片,通过重注释而获得lncRNA的表达谱或者将mRNA进行重注释到新版基因组上。
流程图如下:
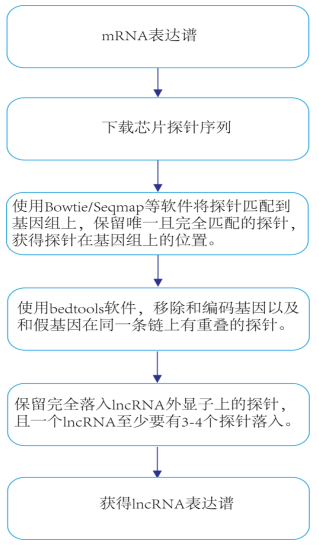
首先是前期准备工作,必要的软件和数据选择(本次以lncRNA为主):
1)Seqmap这个软件是本流程的核心,windows和linux两个版本都有,如果是windows版本需要再DOS下运行(期待后续sangerbox加入此软件);
2)Bedtools这个软件,是处理基因组信息分析的强大工具集合,如何安装使用谷歌一下就能查到,参考;
3)常见的可以重注释大量lncRNA的芯片平台有HG-U133_Plus_2、HuEx、HG-U133A_2。
接下来具体操作步骤:
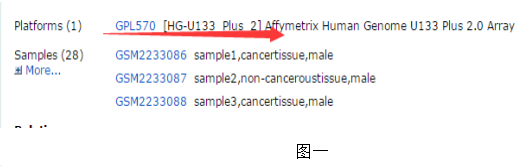
1)从GEO数据库下载想要重注释的芯片平台数据,比如我现在想重注释GSE84402这个芯片数据,跳转到GEO数据库GSE84402,下拉看到它的平台信息(图1)GPL570是HG-U133_Plus_2平台,可以重注释,点击GPL570;
下拉看到web link点击(图二);
 跳转到affemetrix公司页面(图三)如果没有注册需要注册才能下载平台数据;
跳转到affemetrix公司页面(图三)如果没有注册需要注册才能下载平台数据;
 找到HG-U133_Plus_2的fasta格式数据(图四);
找到HG-U133_Plus_2的fasta格式数据(图四);
 如果有对应注释版本的bed文件,就下bed文件(图五);
如果有对应注释版本的bed文件,就下bed文件(图五);
2)芯片平台数据下载完毕,如果下的是Fasta格式数据,需要从UCSC或Ensemble或NCBI数据库中下载对应版本的基因组的Fasta数据(图六、七,以UCSC数据库为例);
 但是如果是已经比对好的bed格式数据则可以直接跳转到下一步(忽略此过程);
但是如果是已经比对好的bed格式数据则可以直接跳转到下一步(忽略此过程);
否则需要使用seqmap软件进行比对,比对要求即探针序列唯一匹配且不允许错配,从而得到探针序列的基因组信息。
最后从GENCODE数据库下载基因组上的注释数据(图八),使用perl或其他方法提取lncRNA、mRNA和假基因的注释信息(bed格式)。同理也可以下载fasta数据,然后使用seqmap比对到基因组上,得到对应的基因组位置。

3)经过以上步骤就获得了探针和lncRNA、mRNA和假基因在基因组上的位置信息。接下来采用bedtools中的intersect命令,得到落入到lncRNA、mRNA和假基因的探针。
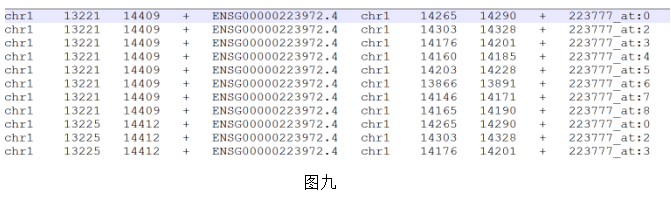
Bedtools命令如下:命令放前面的是在染色体位置长的,后面的是短的,意思是如果你的平台的探针是长的则前面的就是探针的bed,后面的就是假基因的bed。bedtools匹配时没有考虑到正负链,需要用perl提取正负链一致的数据。命令已备好 ./intersectBed -wa -wb -F 1.0 -b /home/Downloads/pseudogene.bed -a /home//Downloads/HG-U133_Plus_2.bed >/home/Downloads/HG-U133_Plus_2_pseudogene.txt,结果如图9
4)最后使用R、excel等软件,获得去除落入到假基因和mRNA的探针,找到唯一落入lncRNA外显子区域的探针,同时要满足每个lncRNA至少要有四个探针落入。
5)将落入到lncRNA的外显子的探针的表达加和即为lncRNA的表达,就这样我们就获得了为自己所用的lncRNA表达谱。
结束语
今天的每周分享就是这么多,下周我们继续相约,期待您的关注!!!

- 发表于 2018-08-05 12:06
- 阅读 ( 8825 )
- 分类:综述
