GSEA 富集分析简述
GSEA定义:
Gene Set Enrichment Analysis (基因集富集分析)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一个是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),一个是表达矩阵,软件会对基因根据其于表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
更多可以查看:
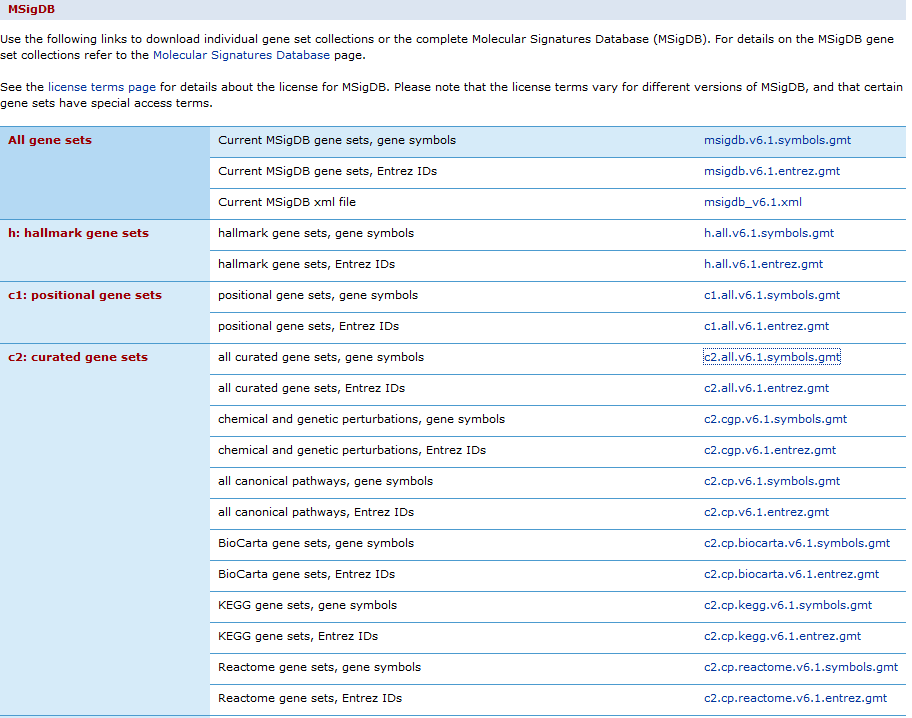
GSEA中几个关键概念:
计算富集得分 (ES, enrichment score). ES反应基因集成员s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度)是相关的。富集得分ES最后定义为最大的峰值。正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。
评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算p-value。
多重假设检验矫正。首先对每个基因子集s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值)
Leading-edge subset,对富集得分贡献最大的基因成员。
- 发表于 2018-08-14 15:07
- 阅读 ( 19956 )
- 分类:软件工具
