生信文章解析(第二篇)使用深度学习的方法整合多组学数据预测肝癌预后
今天要为大家分享一篇去年三月发表在Clinical Cancer Research 的文章[Deep Learning based multi-omics integration robustly predicts survival in liver cancer; 2018.3; IF:9.619]

目前,缺少整合多个样本群的多组学数据预测HCC的生存的研究工作,而识别肝细胞癌(HCC)鲁棒性的生存亚型能够显著改善患者的治疗。所以为了填补这一空缺,本文作者基于HCC提出了一个深度学习(DL)模型,能够将六个样本群的患者鲁棒性的划分为不同的生存亚群。作者使用来自TCGA的RNA-seq,miRNA-seq和甲基化数据,对360 例HCC患者构建基于DL的生存敏感模型,其预测预后的效果不亚于考虑基因组学及临床数据的替代模型。

研究涉及了6个样本群
① TCGA 数据集:来自TCGA的360 例样本的RNA-seq 数据, miRNA-seq 数据, DNA甲基化数据以及临床信息。
② 验证集1(LIRI-JP cohort, RNA-seq):来自ICGC的230 例样本的 RNA-seq 数据 。
③ 验证集2(NCI cohort, microarray gene expression):来自GSE14520的221 例具有生存信息的样本。
④ 验证集3(Chinese cohort, miRNA expression array):来自GSE31384的166对HCC与正常组织的配对样本。
⑤ 验证集4(E-TABM-36, gene expression microarray):来自Affymetrix HG-U133A GeneChips平台的40例具有临床信息的HCC样本 。
验证集5(Hawaiian cohort, DNA Methylation array):来自Illumina HumanMethylation450 BeadChip 平台的27 例具有全基因组甲基化谱的样本。

1.使用机器学习转换特征
2.特征选择以及K均值聚类
3.数据分割以及鲁棒性评估
4.有监督的分类
5.评估模型的指标
6.功能分析

1. 在TCGA样本群中结合HCC的多组学数据识别出两类生存亚型
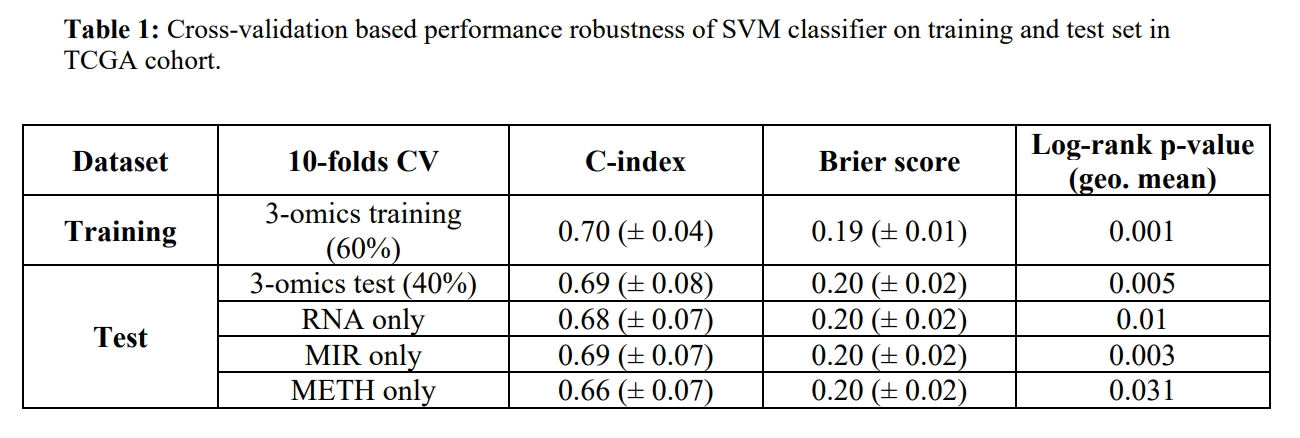 表1
表1
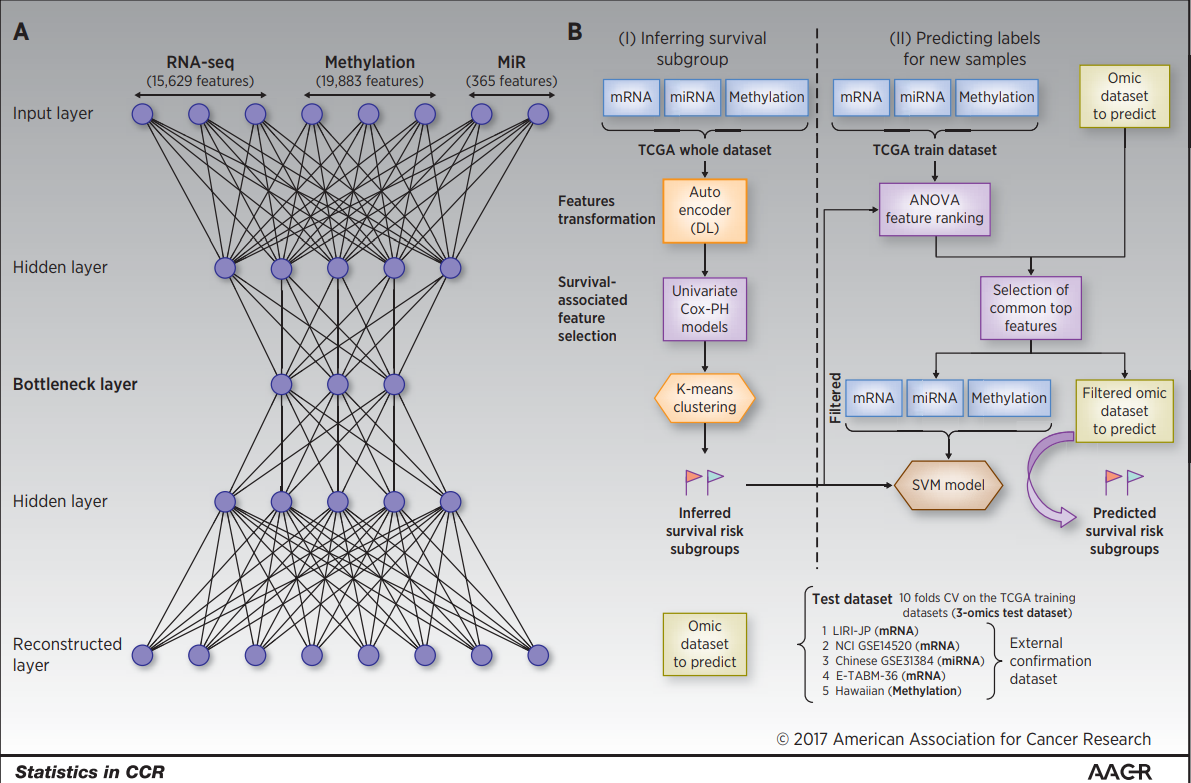 图1
图1
对于TCGA数据,得到基因RNAseq以及miRNA和甲基化数据作为输入特征。使用深度学习方法将这三类组学数据整合到一起,结构如图1A所示。得到100个特征,然后我对100个特征进行单变量Cox-PH回归,发现与生存相关的37个特征。对这37个特征进行K均值聚类,最终确定最优的K是2,将两类展示不同的标签。对这两类使用交叉证实的支持向量机(SVM),具体步骤如图1B。最终如表1所示,训练集数据的 C-index 高,brier score低,并且对生存差异的log-rank的p值显著。这些结果表明,使用聚类标签的分类模型对生存特定的聚类具有较强的鲁棒性。
2.生存亚型在五个独立的数据集中得到了鲁棒性验证
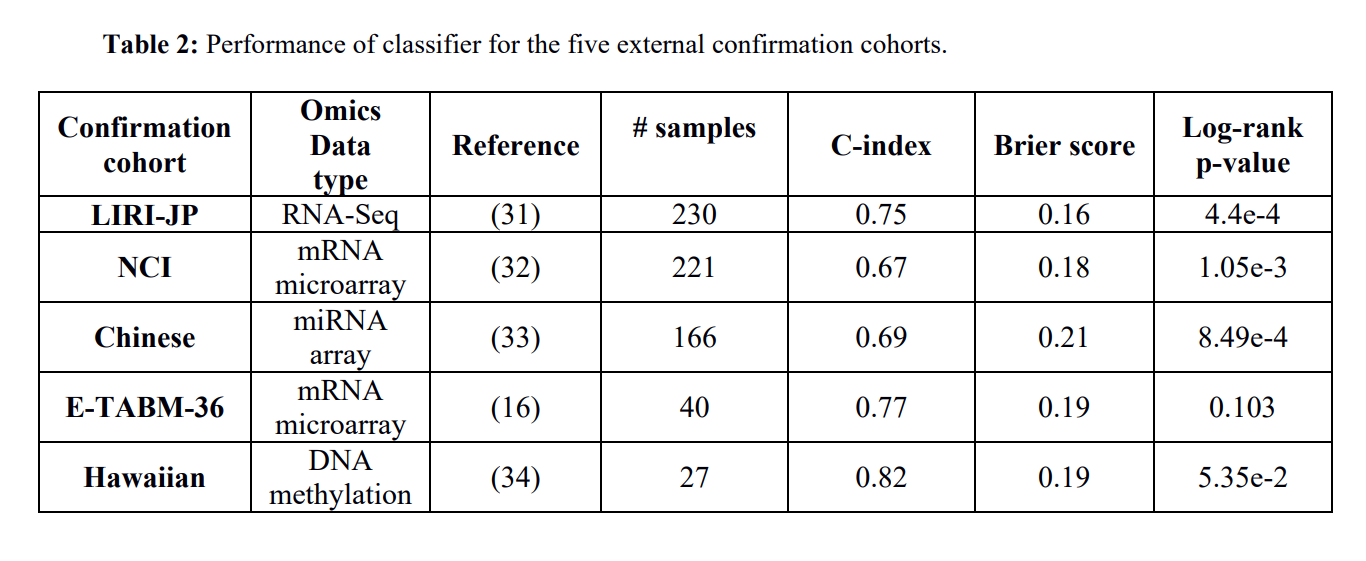 表2
表2
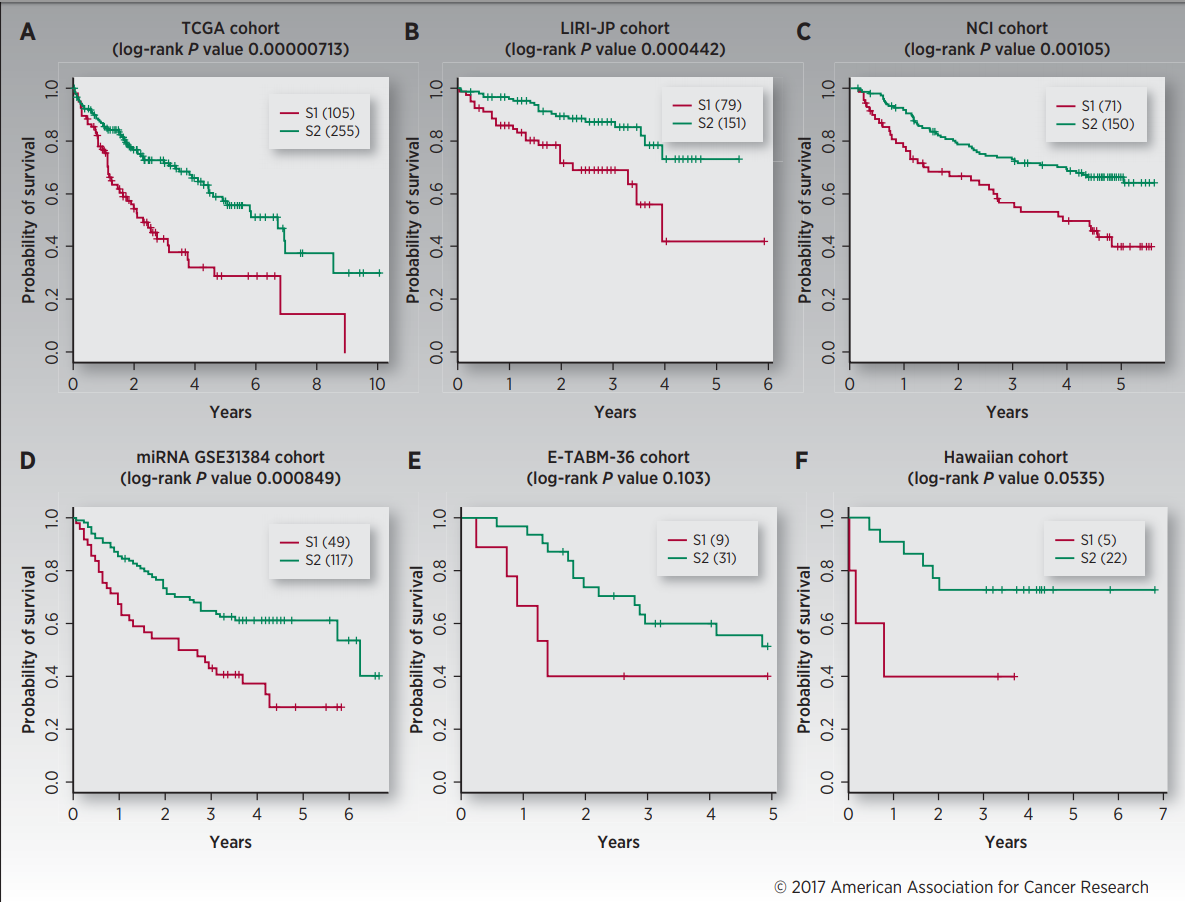
图2
在这一部分,为了验证分类模型在预测生存结局的鲁棒性,作者使用五个独立的样本集来验证,每个验证集的结果如图2所示。接下来作者又对每个验证集评估了指数,可以看到在表2中列出了这五个样本群的C-index,brier score,以及log-rank的p值。
3.DL算法的性能优于其他算法
在这一部分,作者将DL方法与两个其他方法相比较。在第一种方法中,使用传统的降维方法主成分分析(PCA)代替,获得了前100个主成分,然后进行单变量Cox-PH,最终得到13个主要组成分。但是,这种方法在检测生存亚组中给出的log-rank p值并不显著(P = 0.14)。在第二个比较方法中,有37个特征,但是最终得到的log-rank p值仍然不显著。此外,这两类方法在所有验证集中都没有很好的识别出生存亚组。
4. 增加临床信息并不能改善基于多组学数据的DL模型
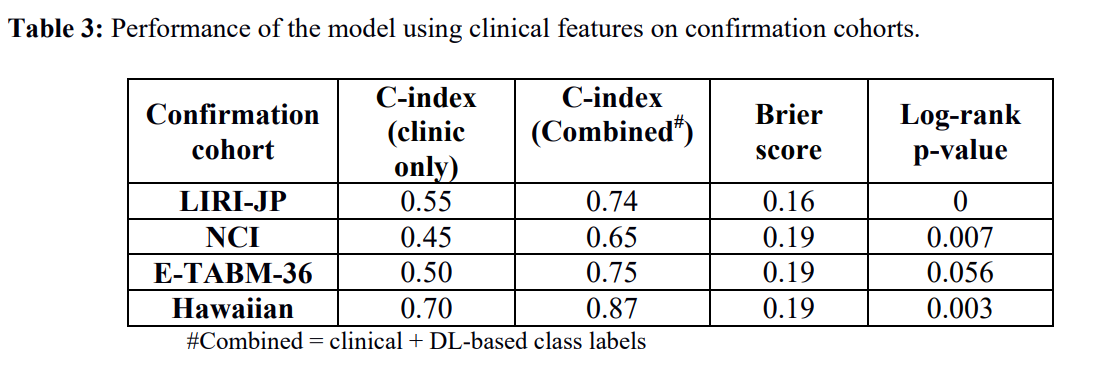 表3
表3
在这一部分作者想探究加入临床信息会不会对模型起到优化效果。因此作者加入临床信息作为特征,对模型进行了评估,评估结果如表3所示。可以看出当临床因素作为特征时,对每个指数进行比较,与未加入临床特征的模型相比较,整体效果并不好。推测其原因可能是DL神经网络的独特优势,它可以通过相关的基因组特征为降低临床特征的冗余性做贡献。
5. 生存亚组与临床因素的相关性
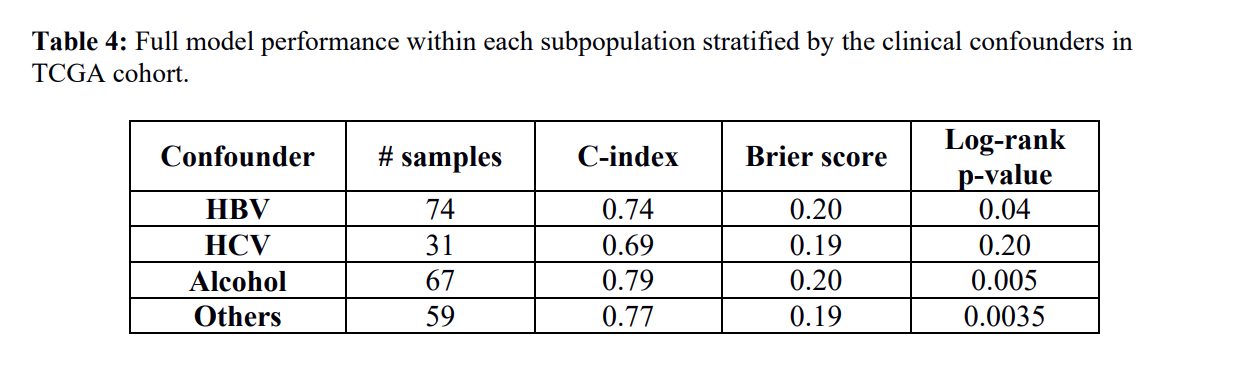 表4
表4
作者在两个生存亚组和临床变量之间进行Fisher 's精确检验,发现只有grade(P=0.0004)和stage(P=0.002)与生存显著相关。由于HCC包括HBC、HCV和酒精在内的多种危险因素,作者就在按个体危险因素分层的样本群中测试了DL的模型(表4),结果表明在多数分级样本群中,模型的效果都比较好。并且TP53突变已经被证实与HCC的预后显著相关,而在这两个亚组中,Fisher 's精确检验结果表明TP53突变具有显著差异。
6. TCGA HCC生存亚群的功能分析
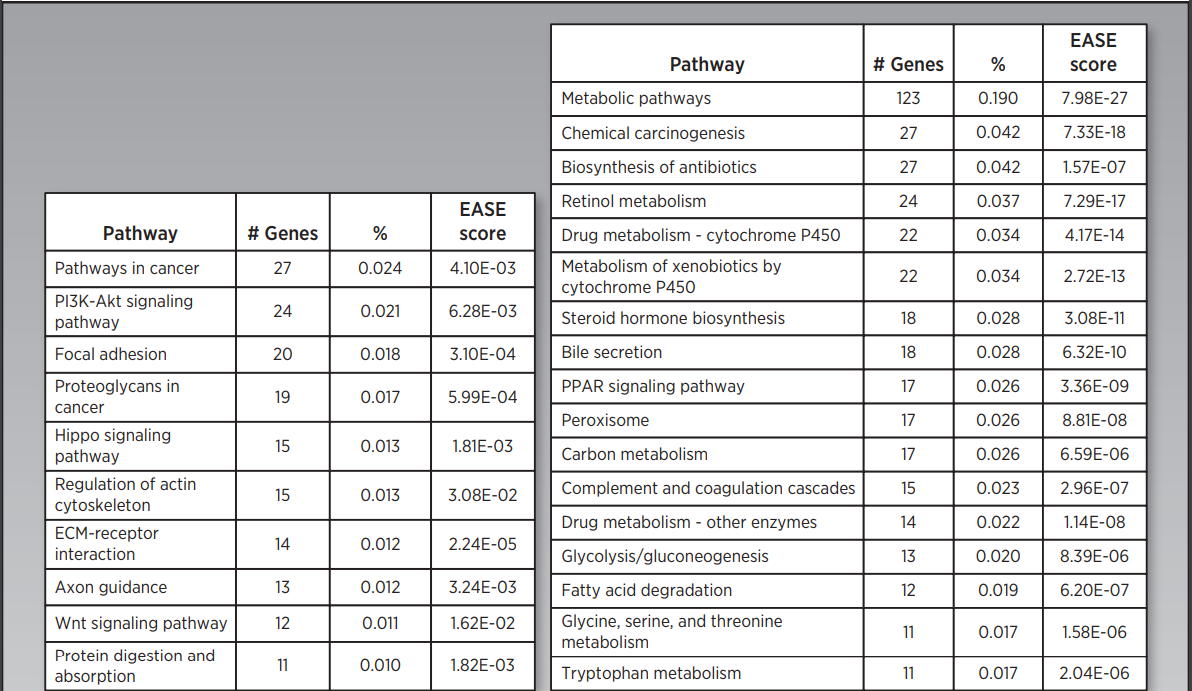
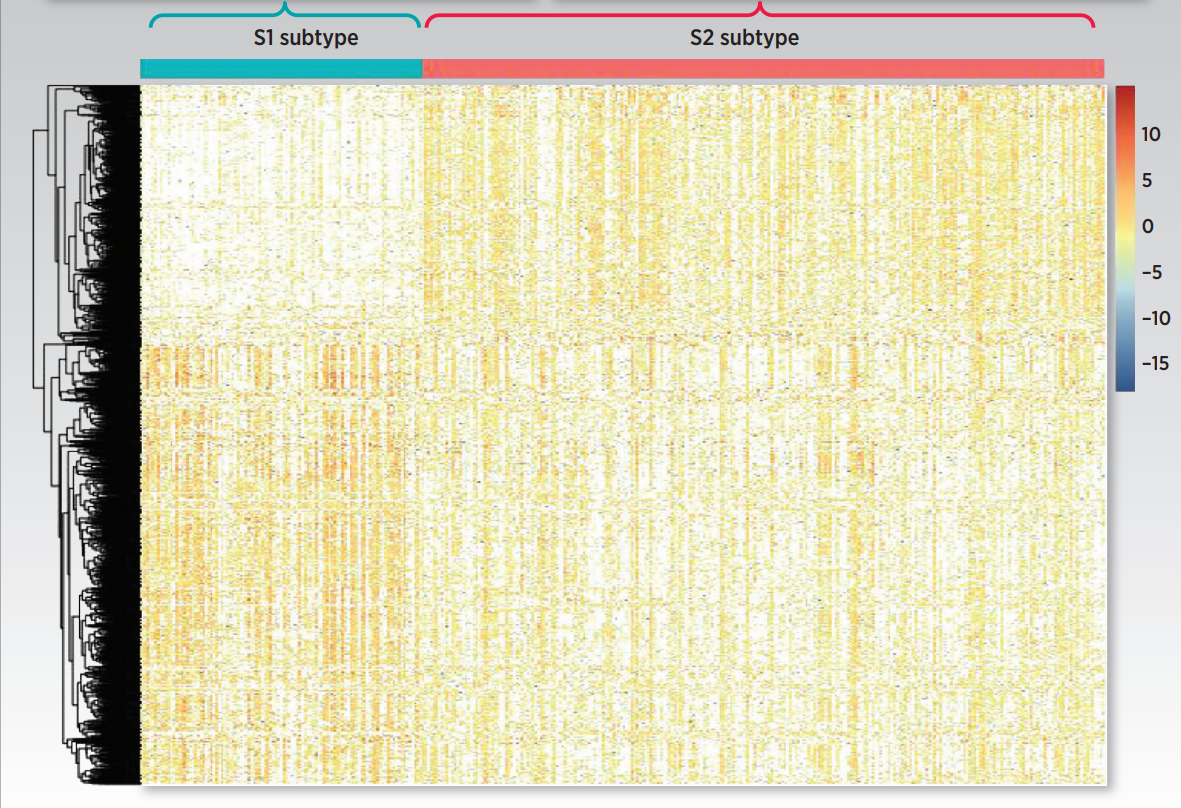
图3
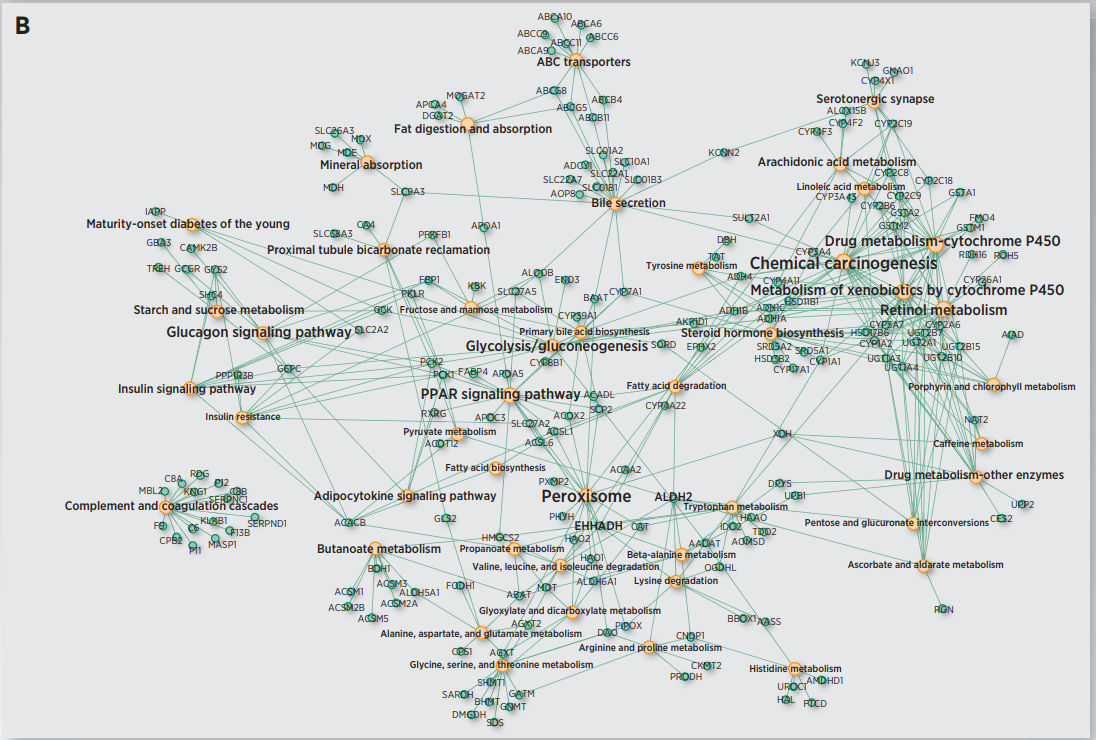
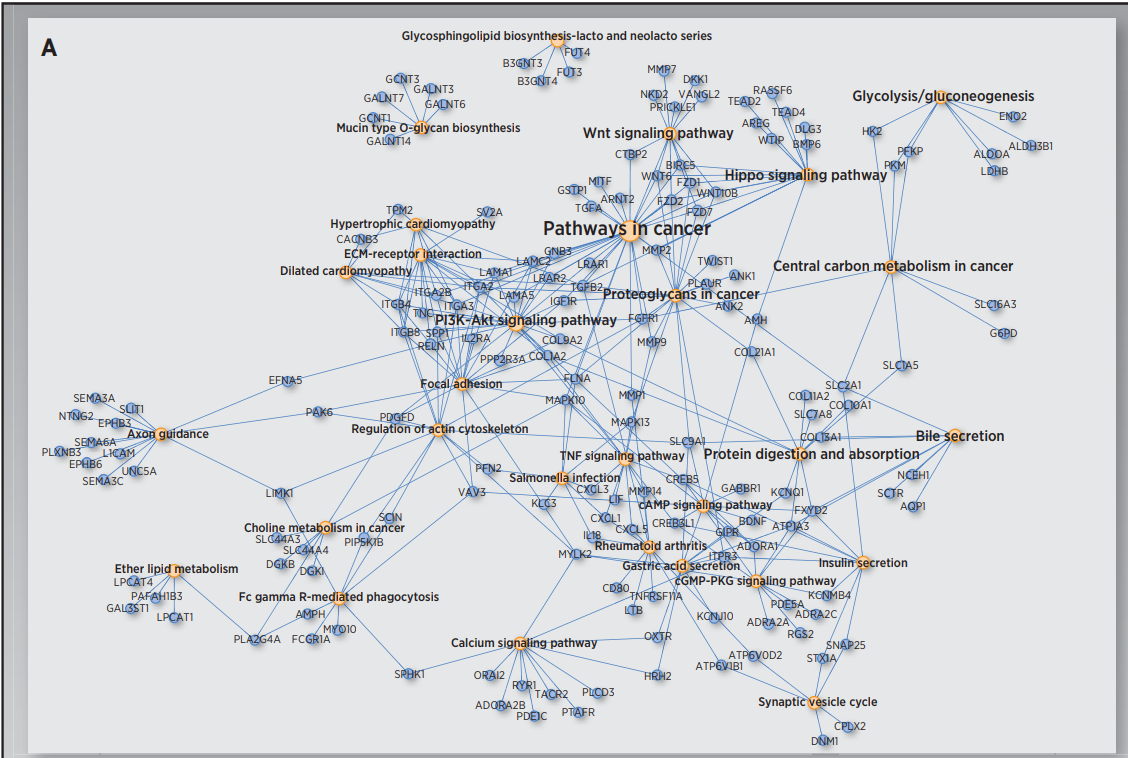 图4
图4
作者首先对识别到的两个生存亚组进行差异表达分析,得到上调基因以及下调基因。图3展示了标准化后的整体的表达信息。这些差异基因包括干性marker基因,癌症marker基因以及已经被证实和HCC进展有关的基因。接下来作者又对这些差异基因进行功能富集,功能富集结果如图4所示。图4A是S1类的富集结果,而S2类的富集结果展示在图4B中。
总结一下,作者使用深度学习的方法整合多组学数据,对HCC样本群进行了预后分型,并且评估了模型的鲁棒性,及分型效能,对这个方法感兴趣或者也在研究预后的小伙伴可以仔细研读下这篇文献哦。

生信文章解析(第一篇)在乳腺癌中,MAPK通路的突变驱动扰动与肿瘤内免疫反应负相关?
生信文章解析(第三篇)生物信息学分析膀胱癌不同亚型中免疫调节基因
生信文章解析(第四篇)能不能找到与PTC分化相关的代谢基因,且进一步识别和PTC预后相关的代谢基因?
生信文章解析(第五篇)IPM对HCC免疫微环境影响的综合分析
- 发表于 2019-07-30 10:55
- 阅读 ( 9408 )
- 分类:文献解读
