生信文章解析(第一篇)在乳腺癌中,MAPK通路的突变驱动扰动与肿瘤内免疫反应负相关?
免疫治疗作为一种新的癌症治疗方法,已经被用于几种癌症的临床治疗。但是,免疫治疗对乳腺癌的治疗有限。由于缺乏对免疫反应基因组特征的认知,这导致无法开发一种新的、有效的治疗乳腺癌的方式。在此,为大家介绍一篇发在ONCOIMMUNOLOGY 上的关于免疫的文献【Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis 2016.10.22 IF:5.333 通讯作者:Michele Ceccarellib & Davide Bedognetti 邮箱:mceccarelli@qf.org.qa dbedognetti@sidra.org 通讯作者单位:多哈Sidra医学研究中心】

在本次研究中,Wouter Hendrickx等人为克服这个缺陷,他们整合TCGA数据库中乳腺癌病人的拷贝数(CNV)、体细胞突变(somatic mutation)以及基因表达谱数据(RNA-seq),首先,基于1004个乳腺癌病人的RNA-seq表达谱数据,通过之前研究报道与免疫治疗耐药相关的转录本的表达将这些样本划分成具有不同免疫表型的样本,即ICR1、ICR2、ICR3和ICR1,其中,在ICR1表型中这些和病人好的预后相关的转录本呈现了高表达(如:PDL1,PD1,FOXP3,IDO1以及CTLA4等)。为说明这种表型刻画的鲁棒性,Wouter Hendrickx等人在另一套包含1954个乳腺癌病人的表达谱数据中去验证他们的这种表型划分标准,同时,Wouter Hendrickx等人又进一步结合拷贝数据去刻画各表型病人特征,他们发现:(1)在ICR4表型病人中Th-1趋化因子CXCL9-11所位于的染色体片段(4q21)发生了显著的扩增;(2)在ICR4-ICR1表型病人中,突变和新生抗原负荷的逐渐减少;(3)在ICR4表型病人中显著发生TP53突变;(4)MAP3K1和MAP3K4的突变和ICR1表型病人相关;(5)在TCGA数据集以及验证数据集中,可以从MAPK的失调的程度去划分乳腺癌病人。他们的研究表明:在乳腺癌中,MAPK通路的突变驱动扰动与肿瘤内免疫反应负相关,通过多MAPK通路的调节可以增强乳腺癌免疫敏感性。
1. 数据的获取:
(1) 训练集:通过R包TCGAbioloinks下载TCGA乳腺癌数据的RNA-seq、临床数据和拷贝数SNP数据,TCGA portal中下载突变数据。首先,需剔除男性样本、组织分期不明确的样本、用过辅助化疗药物的样本以及划分为其他恶性肿瘤的样本(剔除样本数分别为:3, 9, 1, 13, 66, 12)。
(2) 测试集:从GEO数据库、NCI’s caArray数据库以及EMBL-EBI’s ArrayExpress数据库获取那些没有接受辅助化疗药物的原发浸润性乳腺癌芯片表达谱数据(n=1954),包括:raw cel (GSE1456, GSE2034, GSE5327, GSE12093, GSE7390, GSE6532, GSE9195, GSE2603, GSE7378, GSE8193, GSE4922, GSE11121,and GSE45255)、NCI’s caArray database (accession: mille-00271), and the EMBL-EBI’s ArrayExpress database (accession: E-TABM-158),这是数据的平台为:U133A, U133A2, and U133 PLUS 2.0 array platforms。
2. 数据的处理:用R包EDASeq处理TCGA的RNA-seq表达谱数据;对芯片表达谱数据,首先先提取U133A, U133A2和U133 PLUS 2.0平台共有的探针,然后再采用MAS5.0标准化,用COMBAT去批次。
3. 一致性聚类:将20个免疫排斥常数(immunologic constant of rejection,ICR)基因和经典的免疫调控基因映射到TCGA乳腺癌样本表达谱中,ConsensusClusterPlus包对该表达谱进行一致性聚类,最大聚类数设置到7,根据Calinski指数(Calinski index)确定最佳聚类数,进一步刻画这20个ICR基因和经典的免疫调控基因在相应类中的表达情况。
4. 乳腺癌病人中免疫细胞组成的刻画:基于Bindea G等人[1]的工作,得到24个免疫细胞基因集合,采用GSVA包,去计算每个样本中对应的24个免疫细胞基因的ES得分,得到乳腺癌样本的ES值所构成的矩阵,进一步通过层次聚类(聚类方法:Ward.D2),分析上述最佳聚类数下各免疫表型病人间的细胞类组成分布模式,并用gplots绘制热图可视化不同表型病人之间细胞组成的差异。
5. 免疫表型间差异基因的功能富集分析:通过edgeR包计算所关注的免疫表型间的差异基因((log FC) > 0.5 & p<0.05),基于这些显著差异基因的log FC的绝对值排秩,利用GSEA软件实现对这些差异基因功能的刻画,并通过Cytoscape中的插件the Enrichment Map tool可视化。
6. 各免疫表型病人特征分析:进一步分析在各表型病人中stage以及IMS亚型的分布特征,采用circos图可视化这种分布。同时,结合临床数据,通过ggplots2包中的ggkm函数比较各免疫表型病人的生存差异。
7. 突变负荷:基于体细胞突变数据,用NetMHCpan包预测患者特异性HLA I类结合肽。同时,考虑到突变负荷高会产生较多的新抗原,病人的治疗效果会更好,因此,进一步计算各免疫表型病人中非同义突变数的分布是否差异,从而对治疗效果有进一步的评估。
8. 识别与免疫激活相关的突变基因:基于Fisher’s精确检验(Fisher’s exact test)识别表型间的差异基因(p < 0.01),结合MUTSIG分析,从这些差异基因中进一步找出那些高于背景突变频率的基因,将这些基因视为driver gene,并进一步在各表型中刻画这些driver gene。
9. 拷贝数分析:基于TCGA乳腺癌SNP 6.0的数据,采用GISTIC软件去识别样本中相应的扩增和缺失情况,其中,|log 2 ratio| > 0.1是作为缺失和扩增的阈值,只对满足q value < 0.05的扩增和缺失拷贝数进行后续分析。采用NCBI’s Genome DecorationPage(GDP,http://www.ncbi.nlm.nih.gov/genome/tools/gdp)可视化染色体改变情况。
10. MAPK突变得分(MAPK-mut score):首先先将病人分成有MAP3K1/MAP2K4突变的Luminal病人组和无MAP3K1/MAP2K4突变的Luminal病人组,然后从KEGG数据库得到MAPK通路中基因,接下来计算MAPK通路中的基因在这两组病人中的z-score值的差异,得到在两组病人的差异上调的基因和差异下调的基因。先对上下调基因在样本中表达谱的行进行操作,对于每个基因,根据其在样本中的z-score值进行排秩,将基因在样本中的表达谱变成基因在每个样本中表达的排秩情况表达谱;然后对列操作,对于每一个病人,计算基因的秩的平均值,该均值作为样本的MAPK-mut得分。

(1) 各免疫表型的确定:通过ConsensusClusterPlus包对20个ICR基因和经典的免疫调控基因映射到TCGA乳腺癌样本表达谱进行一致性聚类,通过评价Calinski index值(如图Fig 1所示),确定最佳的聚类数为4,划分为:ICR1-ICR4,其中,ICR1 (N = 213)、ICR2 (N = 322)、ICR3 (N = 327)和 ICR4 (N = 142)(如图Fig 2B所示),并发现这20个免疫相关基因在ICR4-ICR1表型病人中整体表达呈现下降趋势(如图Fig 2C所示)
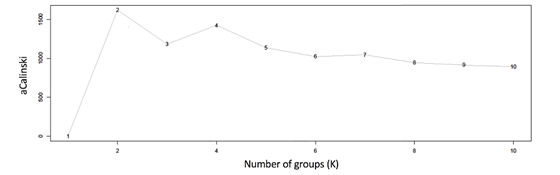 Fig 1. 一致性聚类Calinski index分布图
Fig 1. 一致性聚类Calinski index分布图
 Fig 2.免疫表型的确定
Fig 2.免疫表型的确定
(2) 免疫细胞组成的刻画:基于Bindea G等人[1]整理的24个免疫细胞基因集合,通过GSVA包实现对各表型病人中24种免疫细胞组成的刻画,通过热图的展示发现ICR4和ICR1两个表型的细胞组成有着明显的区别(如图Fig 3所示)。
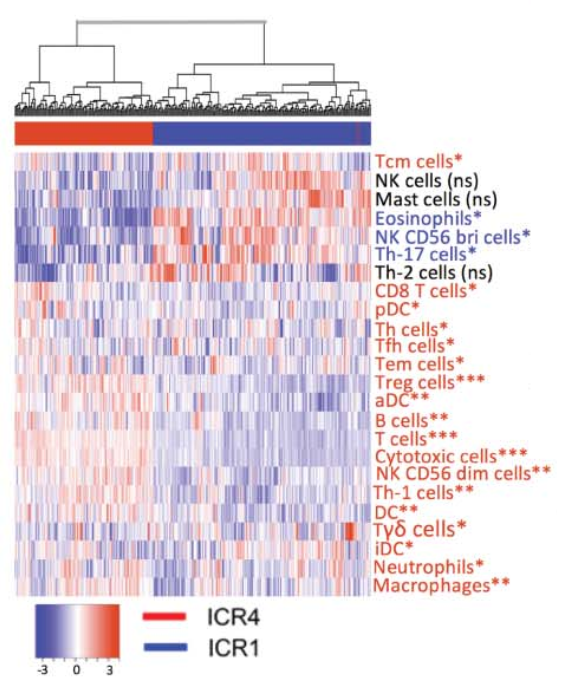
Fig 3.ICR4和ICR1表型病人中24中免疫细胞组成的热图展示
(3) ICR1与ICR4表型病人中差异基因功能富集分析:基Fig 3中ICR4和ICR1表型病人中24中免疫细胞组成差异的现象,分析两个表型病人中的差异基因,并通过GSEA富集这些差异基因的功能,在ICR4表型中,适应免疫相关的通路和功能被激活,如白细胞分化、细胞因子产生、免疫效应过程、B- / T细胞活化以及T细胞增殖和代谢过程(如图 Fig 4所示)。其中,节点表示相应的通路和功能,红色表示在ICR4中高表达基因富集的功能,蓝色表示在ICR4中低表达的基因所富集的功能,线的粗细表示两个功能之间共享基因数目的多少。
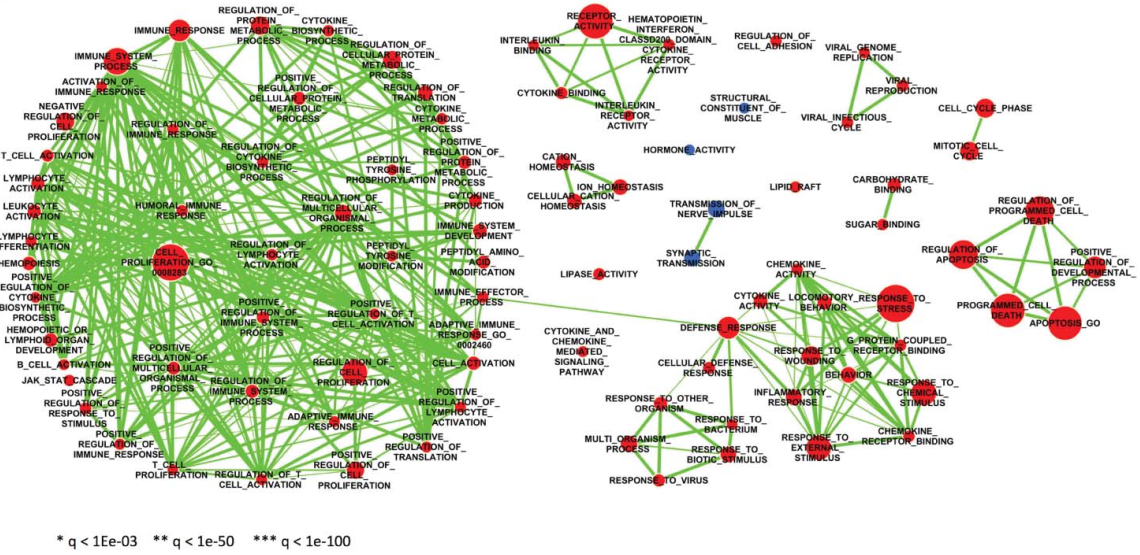
Fig 4. GSEA富集分析
(4)免疫表型病人特征分析:从在各表型病人中stag、IMS亚型的分布特征以及生存情况分析各表型病人的差异(如图Fig 5所示)。
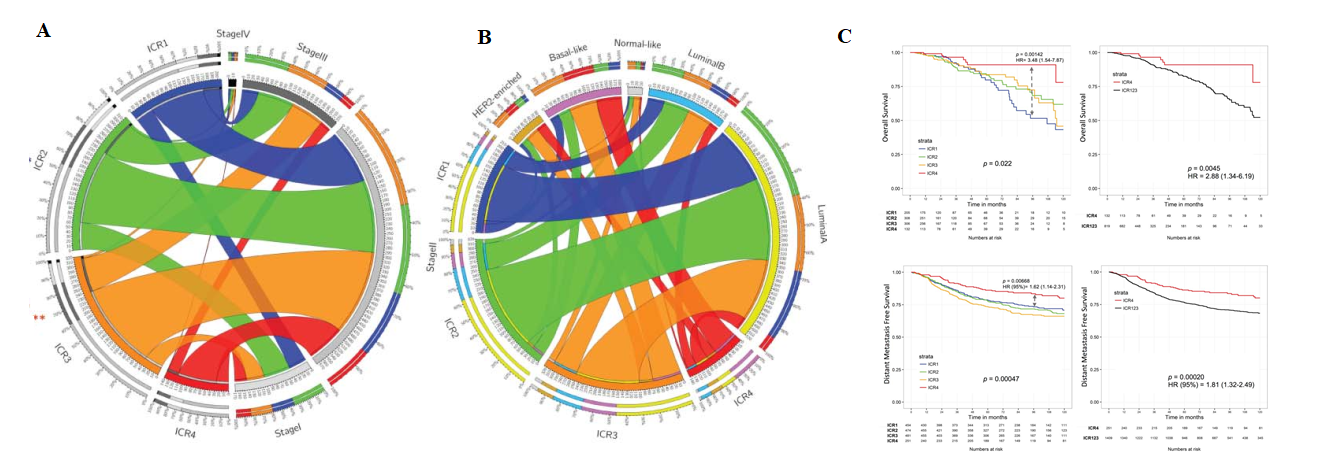 Fig 5. 各免疫表型特征分布.A)stage分布情况;B)IMS亚型分布情况;C)生存情况。
Fig 5. 各免疫表型特征分布.A)stage分布情况;B)IMS亚型分布情况;C)生存情况。
(5) 突变负荷分析:突变负荷高会产生较多的新抗原,病人的治疗效果会更好,因此,结合TCGA病人的突变数据,统计各表型病人中的非同义突变数(如图Fig 6所示),其中,各分类标准下突变数的差异p是通过Kruskal–Wallis test得到的。
 Fig 6. 突变负荷展示.A)non-silent突变数在各不同样本分类标准中的分布情况;B)在样本中的突变率;C)ICR1-ICR4各表型病人突变情况统计。
Fig 6. 突变负荷展示.A)non-silent突变数在各不同样本分类标准中的分布情况;B)在样本中的突变率;C)ICR1-ICR4各表型病人突变情况统计。
(6) driver gene的识别:通过Fisher’s exact test识别ICR1与ICR4表型间的差异基因,共得到64个差异基因(p < 0.01),结合MUTSIG分析,识别出5个显著高于背景突变频率的基因,即:MAP3K1、MAP2K4、TP53、RPGR和POM121,并通过热图、条形图以及boxplot进行在各表型中突变情况进行展示(如图Fig 7所示)。
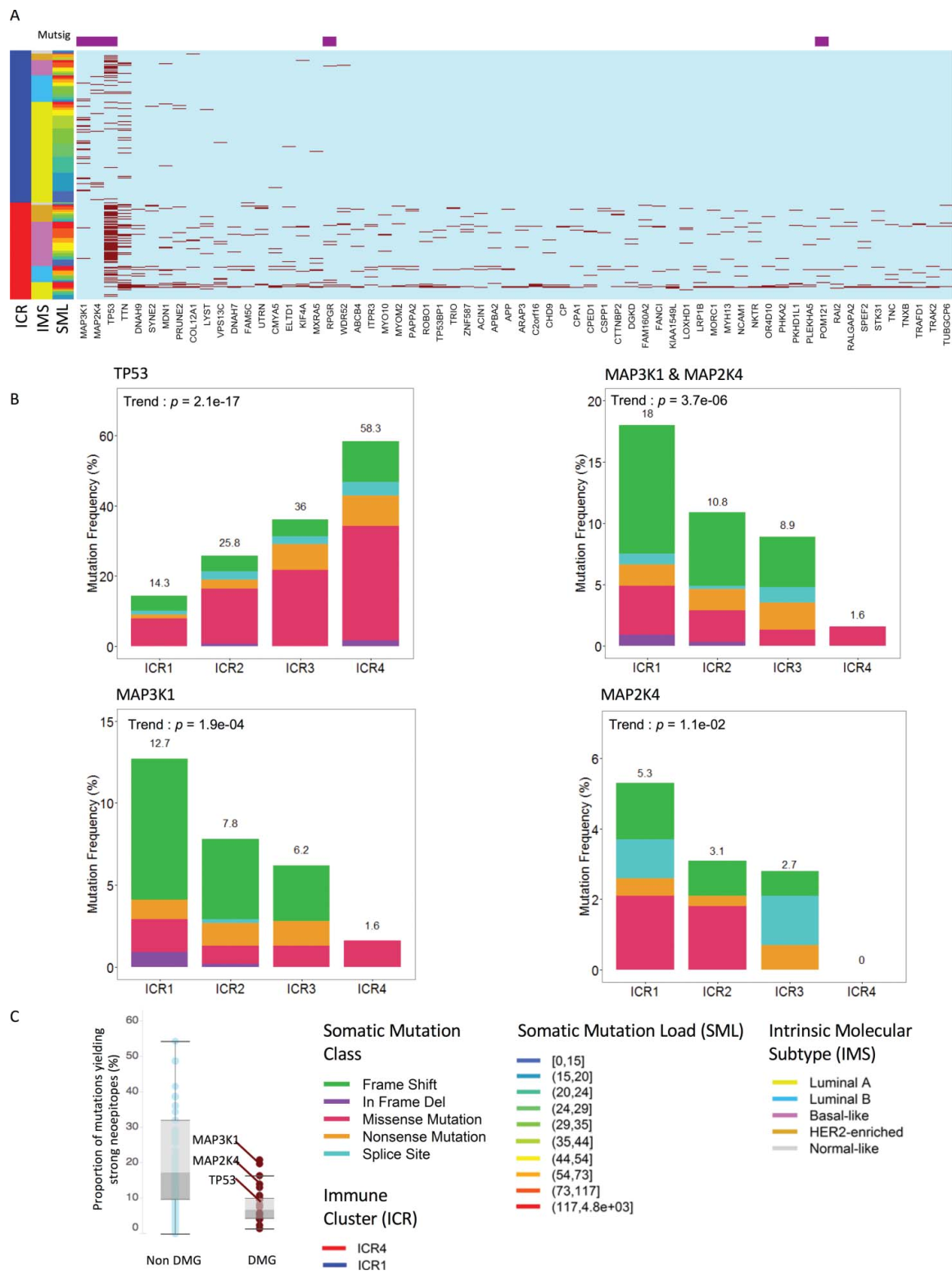 Fig 7. Driver gene的识别和展示
Fig 7. Driver gene的识别和展示
(7) 各表型病人拷贝数情况展示:采用GISTIC软件去识别样本中相应的扩增和缺失情况,通过NCBI’s Genome DecorationPage(GDP,http://www.ncbi.nlm.nih.gov/genome/tools/gdp)对各免疫表型中病人拷贝数改变情况进行可视化(如图Fig 8所示)。
 Fig 8. 各表型拷贝数改变情况的展示
Fig 8. 各表型拷贝数改变情况的展示
(8)通过Fig 7A图,发现MAPK的突变主要发生在luminal乳腺癌病人中,因此,通过整合突变数据和转录组学的数据,想进一步刻画MAP3K1/MAP2K4突变所干扰的功能,向在方法中描述的那样去计算MAPK-mutation得分,发现根据ICR分类,该得分值可以很好的划分luminal样本(如图Fig 9所示),在Fig 10中展示了MAPK通路的失调的通路图,其中红色的五角星表示在ICR4 vs ICR1中上调的基因,绿色的五角星表示在ICR4 vs ICR1中下调的基因。
 Fig 9. MAPK-mutation得分刻画不同免疫表型
Fig 9. MAPK-mutation得分刻画不同免疫表型
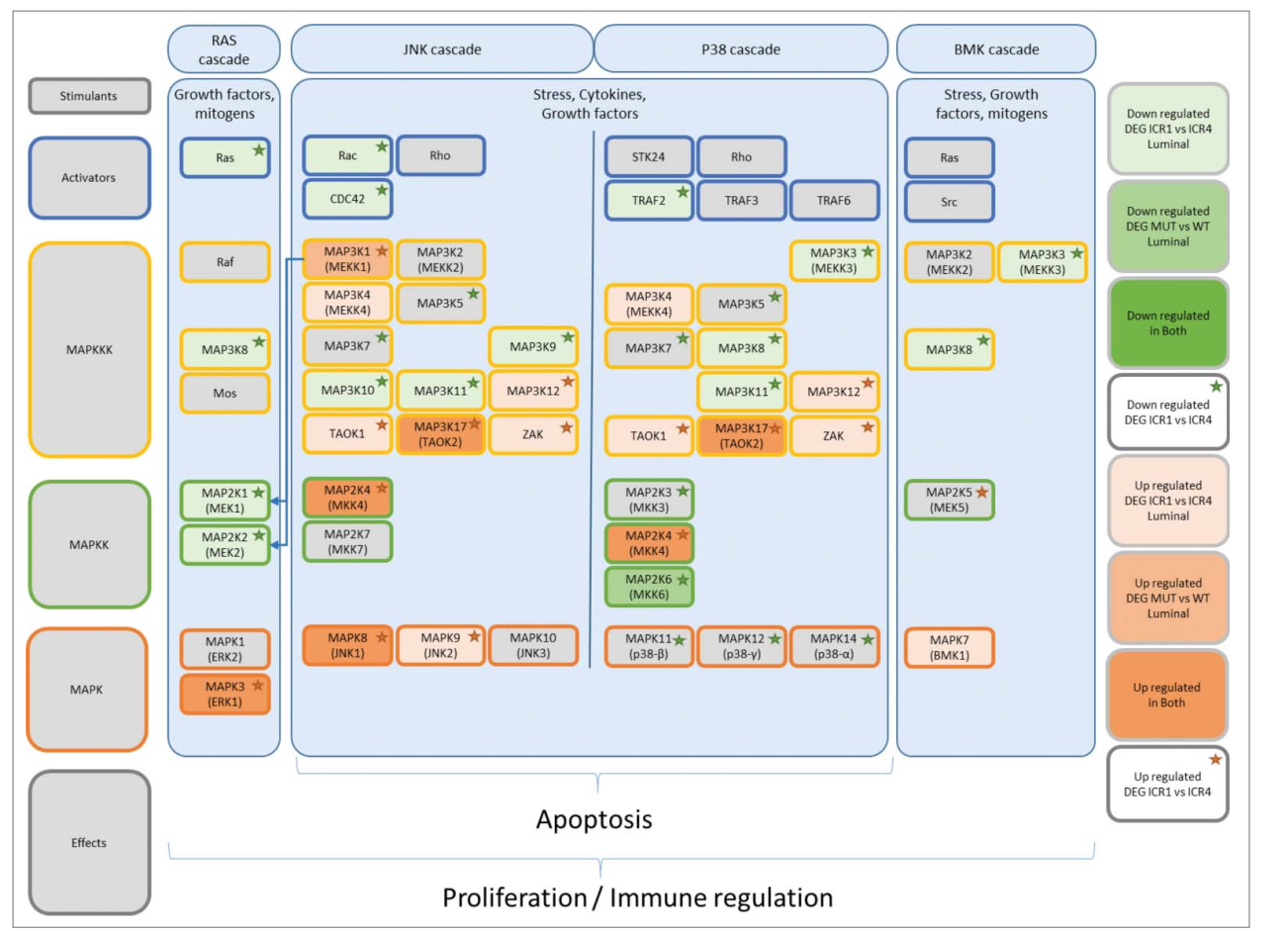 Fig 10. MAPK通路失调示意图
Fig 10. MAPK通路失调示意图
参考文献:
1. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer[J]. Immunity, 2013, 39(4): 782-795.

生信文章解析(第二篇)使用深度学习的方法整合多组学数据预测肝癌预后
生信文章解析(第三篇)生物信息学分析膀胱癌不同亚型中免疫调节基因
生信文章解析(第四篇)能不能找到与PTC分化相关的代谢基因,且进一步识别和PTC预后相关的代谢基因?
生信文章解析(第五篇)IPM对HCC免疫微环境影响的综合分析
- 发表于 2019-07-30 10:21
- 阅读 ( 7539 )
- 分类:文献解读
