介绍几个DNA甲基化差异筛选的软件
DNA甲基化是最早发现的修饰途径之一,可能存在于所有高等生物中。DNA甲基化 导致某些区域DNA构象变化,从而影响了蛋白质与DNA的相互作用,抑制了转录因子与启动区DNA的结合效率,能关闭某些基因的活性,去甲基化则诱导了基因的重新活化和表达。DNA甲基化的主要形式为5-甲基胞嘧啶,N6-甲基腺嘌呤和7-甲基鸟嘌呤。在真核生物中,5-甲基胞嘧啶主要出现在CpG和CpXpG中,原核生物中CCA/TGG和GATC也常被甲基化;没有甲基化的胞嘧啶发生脱氨基作用,就可能被氧化成为U,被DNA修复系统所识别和切除,恢复成C;已经甲基化的胞嘧啶发生脱氨基作用, 它就变为T, 无法被区分。因此, CpG序列极易丢失,甲基化胞嘧啶极易在进化中丢失,所以,高等真核生物中CG序列远远低于其理论值;哺乳类基因组中约存在4万个CG islands,大多位于转录单元的5'区。
近年来随着测序技术的发展,全基因组甲基化测序技术也有了很大的发展(14年之前基本上很少见相关甲基化测序的公司),比较流行的还是WGBS技术,基于WGBS进行全基因组甲基化检测之后进行生物信息分析,一般的分析内容无非是找甲基化位点,甲基化在基因组,染色体,功能元件上的分布,寻找DMS,DMR,DMP,进一步的与基因关联分析相关基因功能。
今天主要来介绍的是两款老少皆宜的筛选甲基化的软件,这两款软件均来自于张岩教授所带领的哈尔滨医科大学计算表观遗传学课题组,张岩教授的课题组从2006年开始就开展研究表观遗传学研究,发表了许许多多的计算表观遗传学相关的算法、软件以及数据库。
首先上一个简单的软件:QDMR: a quantitative method for identification of differentially methylated regions by entropy,这是一个2011年发表在Nucleic Acids Res上的软件
软件界面如下:

从图中可以看出操作及其简单,实际上该软件算法的原理是基于香浓信息商改造而来(比较抽象,学算法者自行百度),就是输入一个甲基化的矩阵,这个矩阵每一行表示一个cpg位点,多种一个基因组区域,每一列表示样本在这些cpg位点或区域的甲基化水平
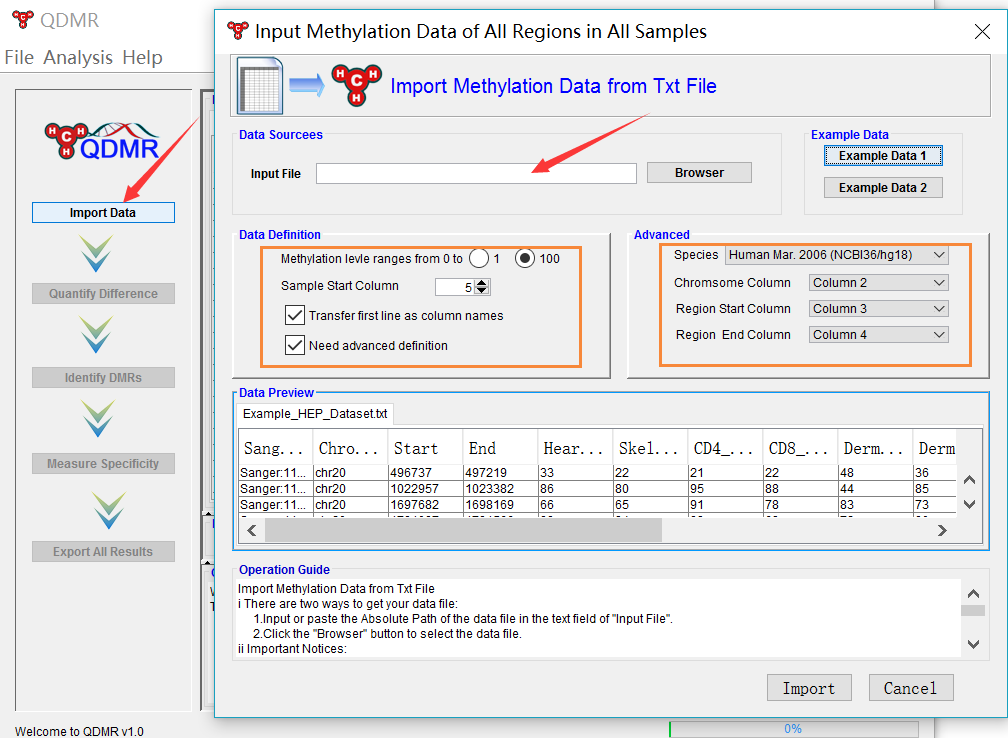
点击Import按钮之后没报错的话数据就导入成功了,进一步点击左侧Quantify Difference按钮进行甲基化差异计算,只需点击,傻瓜式操作,点完之后甲基化差异计算就算完事了,此时选择一个阈值来筛选甲基化差异,点击Identify DMRs,阈值可以根据自己的数据适当调整,一般默认即可,此时就得到了差异甲基化区域及非差异甲基化区域
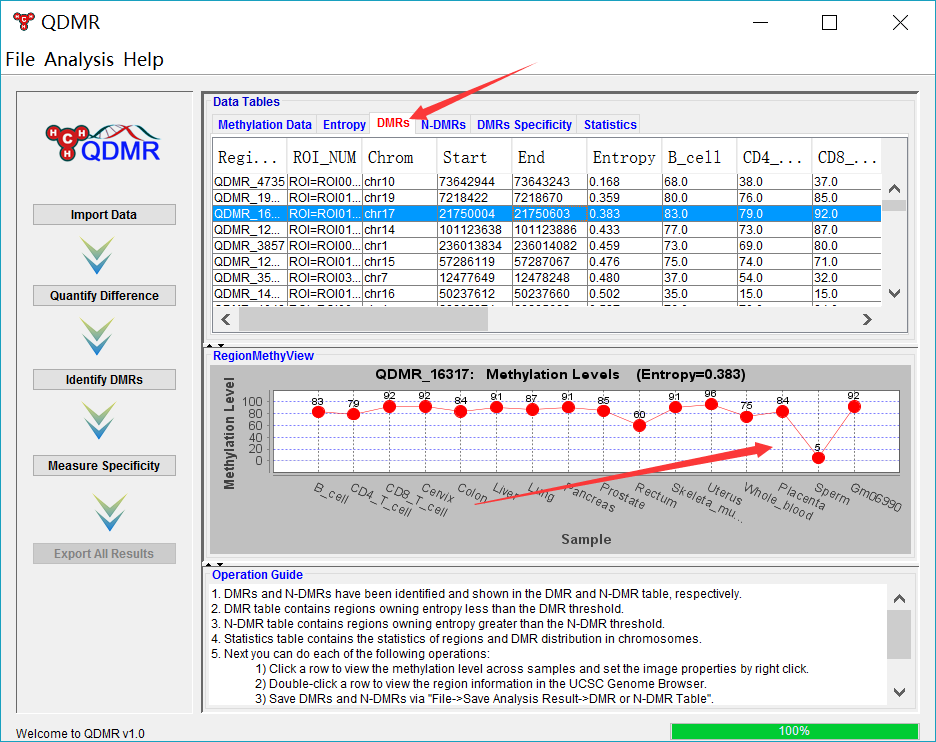
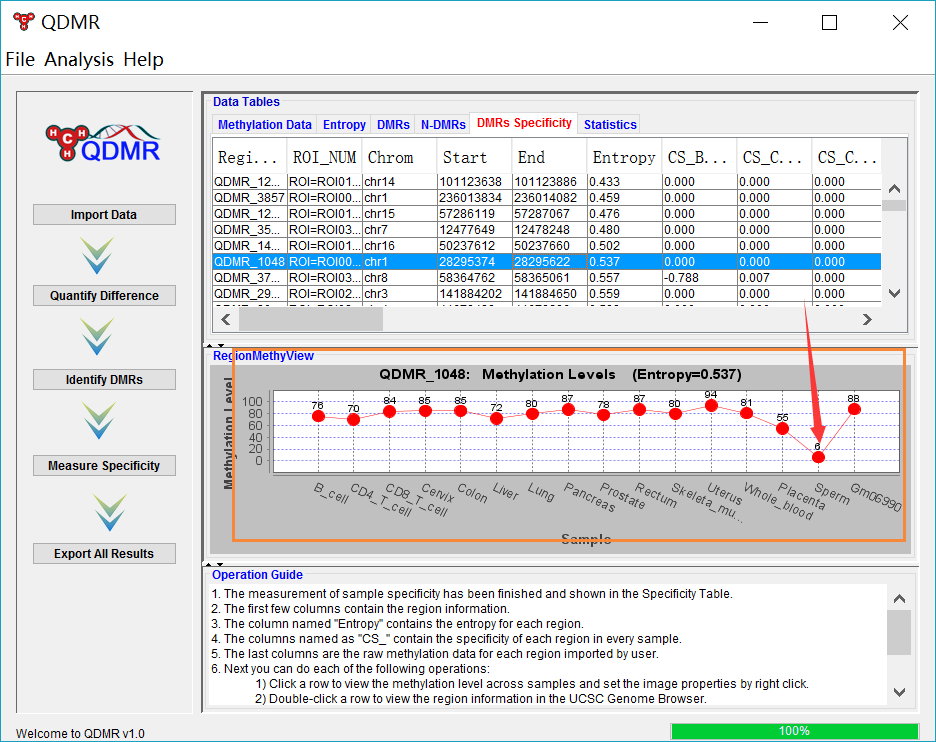
从上面的步骤中细心的同学可以发现软件中筛选的差异甲基化指的是在多个样本中变化剧烈的甲基化区域,而不是相对于某个样本的甲基化,事实上该软件确实只能筛选在多个样本中变化的甲基化区域,筛选出来的差异甲基化区域可以简单的理解为该甲基化区域在多个样本中甲基化水平不稳的。
另外一个问题如果自己的数据有重复样本该如何是好,如果自己的数据有很多甲基化区域并不确定比如BS-Seq数据结果;那么你就需要作者的另外一款软件SMART
SMART:Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes
这是一款2016年发表在 Nucleic Acids Res上的软件包,官网:http://fame.edbc.org/smart/ 安装也超级简单:
pip install SMART-BS-Seq
这款软件是QDMR的改进版,虽然是改进版,其实相当于全新开发,除了都是基于香农信息熵,其他的基本上都改了。
附上工作原理:
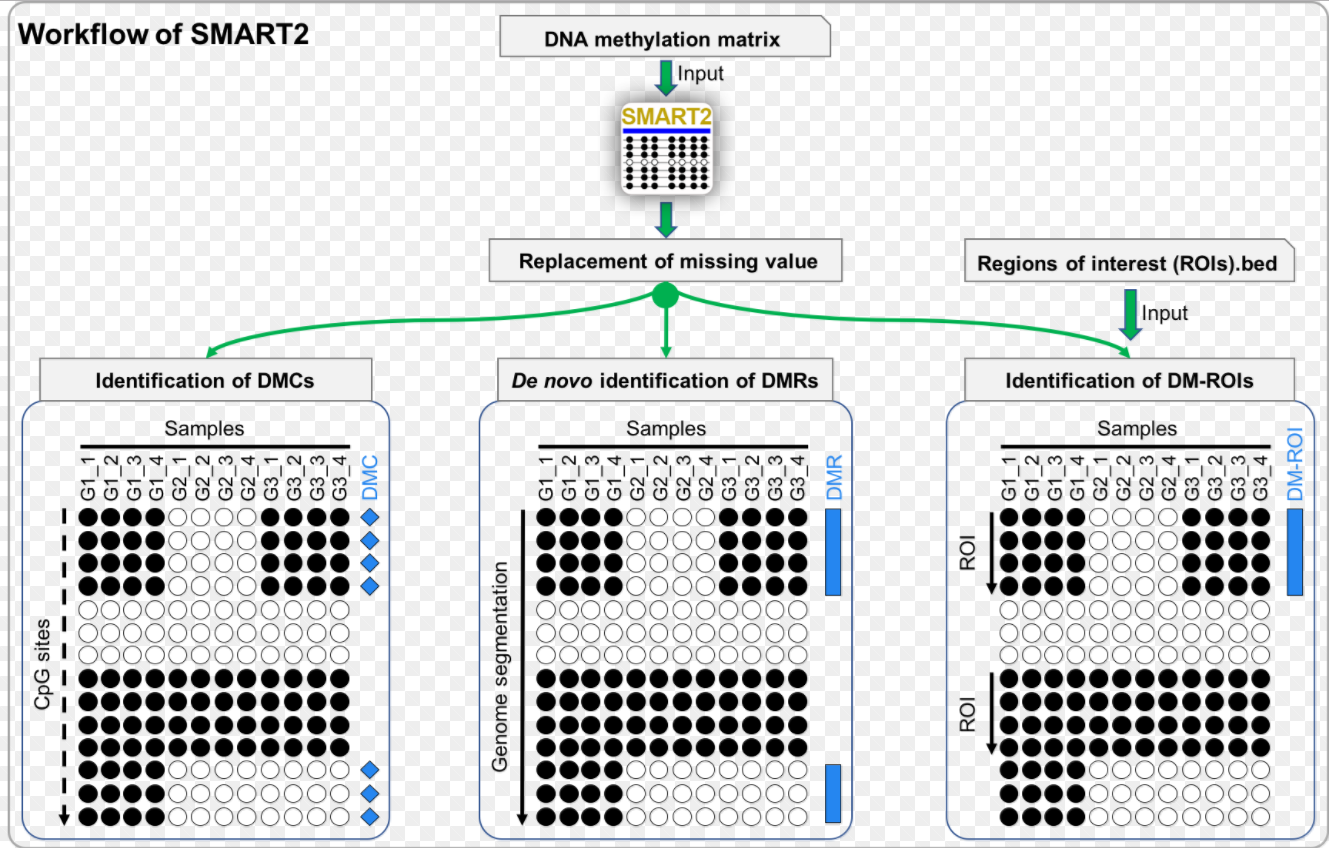
从原理图中可以看出DMCs DMRs DM-ROIs都可以做了
安装完SMART之后初步运行下:
这款SMART非常适合公司在标准流程中使用,谁用谁知道。
引用文献:
Hongbo Liu et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes Nucleic Acids Res: 2016 ,44(1) ,75-94.
Hongbo Liu et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes Nucleic Acids Res: 2016 ,44(1) ,75-94.
最后安利一下软件作者:
刘洪波
哈尔滨医科大学 , 副教授
研究兴趣:Bioinformatics , Computational Biology , Epigenetics
刘洪波,男,博士,副教授。主要研究方向是生物信息学和计算表观遗传学。目前的研究工作主要集中于癌症干细胞的表观遗传调控机制。整合高通量的基因组和表观基因组学数据,基于生物信息学的方法和策略挖掘与癌症发生发现密切相关的表观遗传调控元件,并研究各种调控元件协同调控肿瘤的生成、转移的分子机制。自2007年从事生物信息学研究至今已累计发表SCI论文22篇,总SCI影响因子118.8。通讯/(并列)第一作者论文11篇,其中有6篇SCI论文发表在影响因子为9.112的国际著名期刊《Nucleic Acids Research》上。目前主持国家自然科学基金1项,完成省级、校级课题两项,先后参加国家级、省级课题5项。获得黑龙江省科学技术学术成果奖一等奖2项,于哈尔滨工业大学攻读博士期间获得博士国家奖学金1项。
- 发表于 2017-06-10 17:21
- 阅读 ( 14451 )
- 分类:软件工具
