TCGA CNV全攻略
1、 明白什么是CNV
对正常人来说,基因组应该是二倍体的,所以凡是测到非2倍体的地方都是CNV。但是CNV本身就是人群遗传物质多样性的体现,所以对癌症样本来说,是需要过滤掉正常人体内的germline的CNV,得到somatic的CNV。
CNV(copy-numbervariant)是指拷贝数目变异,也称拷贝数目多态性(copy-number polymorphism,CNP),是一个大小介于1kb至3MB的DNA片段的变异,在人类及动植物基因组中广泛分布,其覆盖的核苷酸总数大大超过单核苷酸多态性(SNP)的总数,极大地丰富了基因组遗传变异的多样性。按照CNV是否致病可分为致病性CNV、非致病性CNV和不明临床意义CNV。
2、TCGA的CNV测量及计算
TCGA里面主要是通过Affymetrix SNP6.0 array这款芯片来测拷贝数变异值得注意的是,并不是只有TCGA利用了SNP6.这个芯片数据,著名的CCLE计划也对一千多细胞系处理了SNP6.0芯片,数据也是可以下载的。
对SNP6.0的拷贝数芯片来说,通常是用PICNIC等软件处理原始数据,就可以得到的segment记录文件,每个样本一个结果,下面是示例结果:
ChromosomeStartEndNum_ProbesSegment_Mean
1617351510801226-0.0397
11627918167260317-0.92
116875871615349781760.0077
116153536161539255-2.7441
116154201161550104-0.8711
11616566172768498346300.0048
1727689167281114846-1.7394
17281190495674710149010.0026
195676511956765182-1.6636
表明了某条染色体的某个区域内,SNP6.0芯片设计了多少个探针,芯片结果的拷贝数值是多少(这个区域的拷贝数用Segment_Mean)。
通常二倍体的Segment_Mean值为0,可以用-0.2和0.2来作为该区域是否缺失或者扩增。
具体数据处理流程见NIH的TCGA官网:
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/CNV_Pipeline/
参考文献:http://mcr.aacrjournals.org/content/12/4/485.long
3、 TCGA的CNV数据下载
众所周知,TCGA的数据的开放程度分成了4个等级,一般人都是下载level 3 的数据,对CNV数据也是如此。
我比较喜欢去broad institute下载TCGA的数据,所有的文件都以目录的形式存放着:
https://gdac.broadinstitute.org/runs/stddata__latest/
https://gdac.broadinstitute.org/runs/analyses__latest/
如果要下载level3的数据,就用``stddata__latest`` 这个url即可,打开可以看到里面列出了所有的癌症种类,假如我们感兴趣的是BRCA,就直接点击进入,用下面的url即可。
https://gdac.broadinstitute.org/runs/analyses__latest/data/BRCA/20160128/
打开url可以看到非常多的文件,这里我们感兴趣的是snp6芯片的拷贝数结果,而且一般是基于hg19版本的。
wget -c -r -np -nH -k -L --cut-dirs 6 -p -A "*snp_6*hg19*Level_3*" http://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/BRCA/20160128/
如果要下载其它癌症种类,只需要改变url里面的BRCA即可。
如果要下载其它类型的数据,只需要改变-A 后面的匹配规则即可,其实就是打开上面url看到的几十个文件的文件名的规律。
"*snp_6*hg19*Level_3*"
几分钟就下载完数据啦,然后你就会看到下面两个截然不同的:
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_hg19__seg
Merge_snp__genome_wide_snp_6__broad_mit_edu__Level_3__segmented_scna_minus_germline_cnv_hg19__seg
其中minus了germline的CNV的就是我们想要的癌症相关的somatic CNV咯!
4、拿到CNV做什么?
首先两个segment文本文件已经可以直接载入IGV查看所有BRCA样本的CNV情况啦,如下所示:
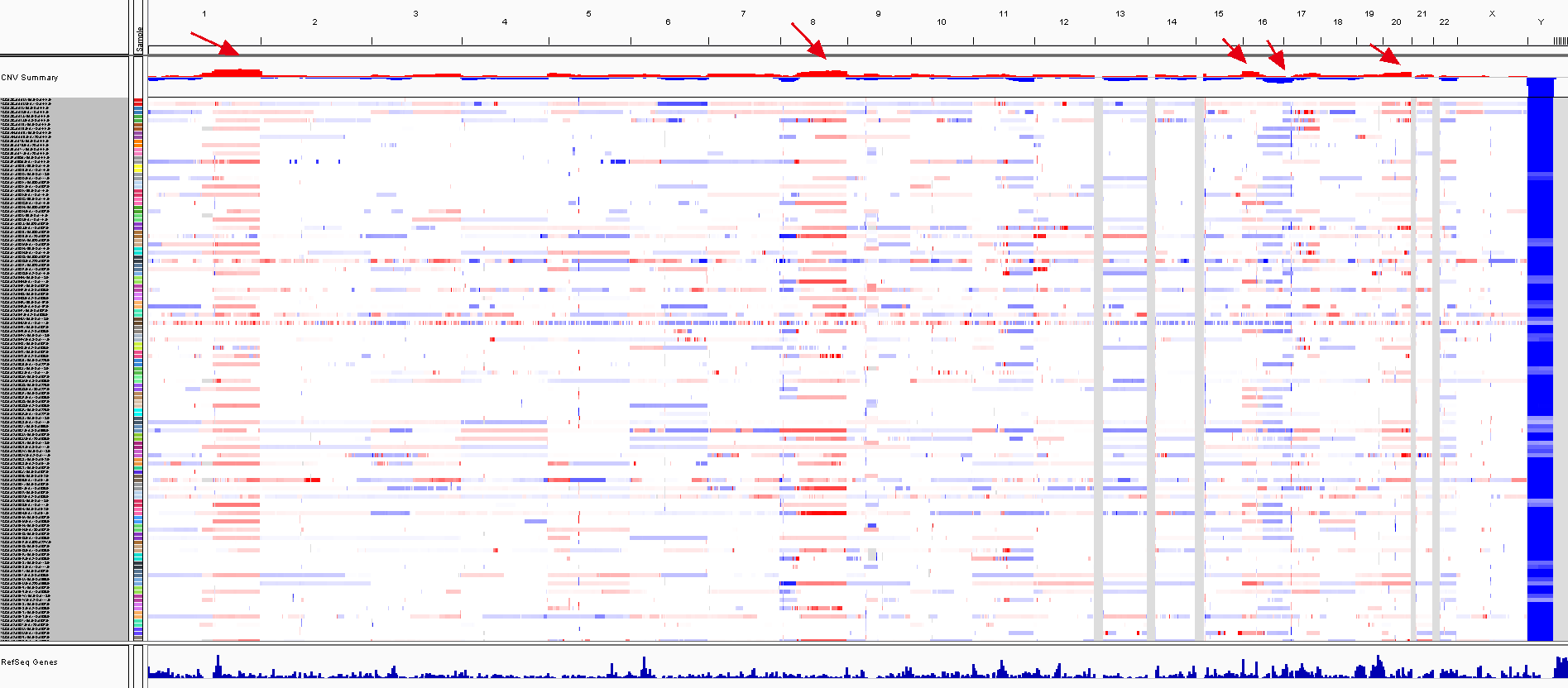
5、 CNV深度分析
1)注释基因
前面我们下载的CNV都是基于基因组区域的,比如1号染色体的61735起始坐标到1510801终止坐标。在IGV里面倒是可以看出一些pattern,但是人们感兴趣的往往是这些位置上面到底有哪些基因。接下来就可以对基因进行各种下游分析。
既然是对基因组片段做基因注释,那么首先就需要拿到基因的坐标信息咯,我是在gencode数据库里面下载,然后解析成下面的bed格式的,如下:
head ~/reference/gtf/gencode/protein_coding.hg19.position
chr16909170008OR4F5
chr1367640368634OR4F29
chr1621096622034OR4F16
chr1859308879961SAMD11
chr1879584894689NOC2L
chr1895967901095KLHL17
chr1901877911245PLEKHN1
chr1910584917473PERM1
chr1934342935552HES4
chr1936518949921ISG15
然后要把我们下载的CNV文本文件,转为bed格式的,就是把列的顺序调换一下:
head Features.bed
chr1321861095674710TCGA-3C-AAAU-10A-01D-A41E-01532250.0055
chr19567651195676518TCGA-3C-AAAU-10A-01D-A41E-012-1.6636
chr195680124167057183TCGA-3C-AAAU-10A-01D-A41E-01248860.0053
chr1167057495167059336TCGA-3C-AAAU-10A-01D-A41E-013-1.0999
chr1167059760181602002TCGA-3C-AAAU-10A-01D-A41E-019213-8e-04
chr1181603120181609567TCGA-3C-AAAU-10A-01D-A41E-016-1.2009
chr1181610685201473647TCGA-3C-AAAU-10A-01D-A41E-01120020.0055
chr1201474400201474544TCGA-3C-AAAU-10A-01D-A41E-012-1.4235
chr1201475220247813706TCGA-3C-AAAU-10A-01D-A41E-0129781-4e-04
避免重复造轮子,我就用bedtools解决这个需求吧,命令很简单,如下:
bedtools intersect -a Features.bed -b ~/reference/gtf/gencode/protein_coding.hg19.position -wa -wb \
bedtools groupby -i - -g 1-4 -c 10 -o collapse
注释结果,我挑了几个可以看的给大家,可以看到,每个CNV片段都注释到了对应的基因,有些特别大的片段,会被注释到非常多的基因。
chr84258492442783715TCGA-5T-A9QA-01A-11D-A41E-01CHRNB3,CHRNA6,THAP1,RNF170,HOOK3
chr84278972842793594TCGA-5T-A9QA-01A-11D-A41E-01HOOK3
chr84279795742933372TCGA-5T-A9QA-01A-11D-A41E-01RP11-598P20.5,FNTA,HOOK3
chr87095267370964372TCGA-5T-A9QA-01A-11D-A41E-01PRDM14
chr104294797043833200TCGA-5T-A9QA-01A-11D-A41E-01BMS1,RET,RASGEF1A,ZNF33B,CSGALNACT2
chr10106384615106473355TCGA-5T-A9QA-01A-11D-A41E-01SORCS3
chr10106478366107298256TCGA-5T-A9QA-01A-11D-A41E-01SORCS3
chr10117457285117457859TCGA-5T-A9QA-01A-11D-A41E-01ATRNL1
chr116899017369277078TCGA-5T-A9QA-01A-11D-A41E-01MYEOV
chr117637870876926535TCGA-5T-A9QA-01A-11D-A41E-01LRRC32,B3GNT6,OMP,TSKU,MYO7A,ACER3,CAPN5
2) 找somatic CNVs

仔细看上面IGV的pattern你会发现某些染色体的某些片段经常会扩增或者缺失,这个现象就是人们想研究是recurrent CNV regions,当然不会用肉眼看咯,这时候需要用GISTIC这个软件。
找到了recurrent CNV regions同样是需要进行基因注释,才能进行下游分析咯。
PICNIC软件用法:http://www.bio-info-trainee.com/1299.html
GISTIC软件用法:http://www.bio-info-trainee.com/1648.html
TCGA : http://www.biotrainee.com/thread-1696-1-1.html
- 发表于 2017-06-16 17:28
- 阅读 ( 19466 )
- 分类:基因组学
