MECAT:PC组装人的基因组
看到这个题目,大多数人的反应,肯定是小编忘吃药了。no no no 小编今天很正常,利用个人PC来组装人的基因组绝对不是痴人说梦。
首先,小编先给大家介绍一些关于组装的历史。大家肯定也知道,这个组装技术的发展是依赖于测序技术的。首先在一代测序的时候,测序数据量较少,成本较高,人们对于组装的结果预期也比较低,弄到contig水平就可以啦,还有就是这个组装的物种基因组都偏小。因此那个时候的组装软件都是基于overlap的。然后呢,二代测序技术来了,数据量超高,成本也便宜啦。科研工作者就想啊,现在都二代了,这个组装的预期得提高啊,得与时俱进啊。但是基于overlap的组装不了太长,而且超级慢,又加之,二代数据量太多,overlap扛不住啊。因此有人就开发出了基于图论的组装算法。然后呢,三代测序技术这两年出来了,科学家一看,我x,这么长的read,那还搞毛图论的算法,直接简单粗暴点,用overlap多简单,于是基于overlap的软件又火啦。
说到这,刚进入问题,简单粗暴的overlap难道就一统天下了吗?难道所谓的优化算法,就真的没有未来了吗?
当然
不是。
今天给大家推荐一款牛叉的三代组装软件,号称资源浪费少,绿色又环保,而且在个人笔记本上毫无压力的软件——MECAT。
赶紧膜拜,预发表文章:
MECAT: an ultra-fast mapping, error correction and de novo assembly tool for single-molecule sequencing reads(肖传乐)
接下来,小编跟大家简单的墨迹下这个文章中的内容。大家简单了解下。
这个三代测序啊,有一个问题,就是错误率有点高,15%。也就是说这个数据啊,不能直接用来组装,你得纠错,怎么纠错呢。做过纯三代组装的人,都知道。是用三代自身去纠最长的read。问题来了,这里如果用测得全部的reads都跟最长的read去比,这一步超级耗时。
这一步,怎么办啊。
有没有相应的算法可以优化下。
如果你能提出这样的问题,小编恭喜你,你这个小伙子还是很有前途的。
肖老师就是构建了一套打分矩阵,使得这里不需要全部都比一次,从而缩短了时间。
当然肖老师,是个工作狂,一看搞下去有前途,还返场优化了下其他部分。
现在比较常用的三代数据组装软件有Canu、Falcon和HGAP等,这些软件在组装之前都是通过两两比对确定overlap,之后再纠错的。这一步时间超级长,举例,PBcR-MHAP软件平均有84%的时间浪费在纠错部分。这一步不仅仅耗时,而且超级占用资源。这里是婶婶也不能忍受的地方。
这个软件牛叉在它的优化算法,算法示意图如下:
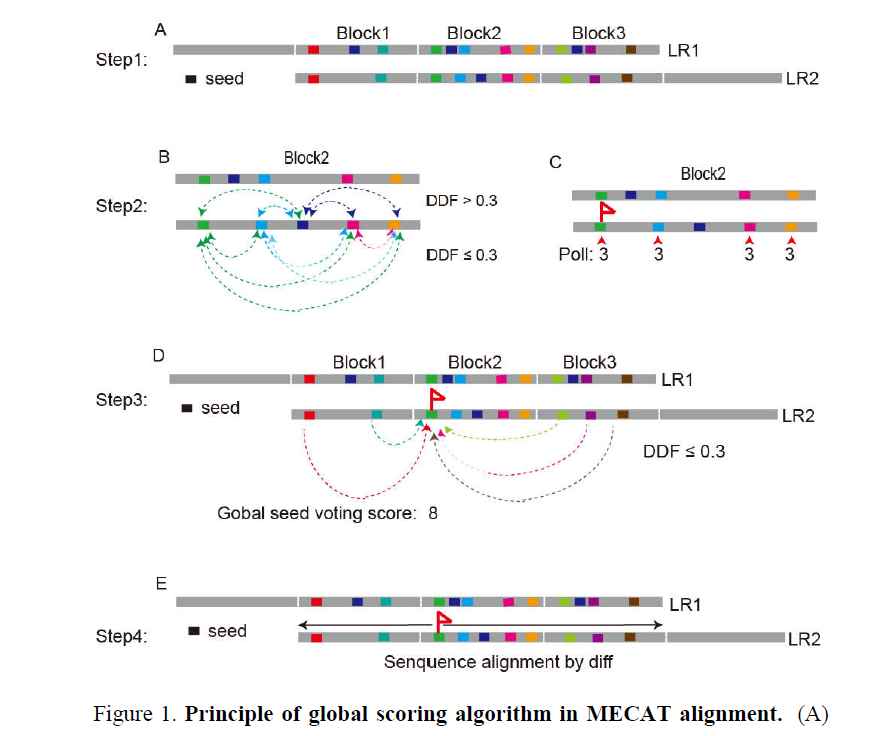
A Reads被分成多个Block;
B. 对所选Kmer pair相互打分;
C. 选取最高得分作为种子;
D. 将种子区域与其他区域进行比较;
E. 最终确定reads关系。
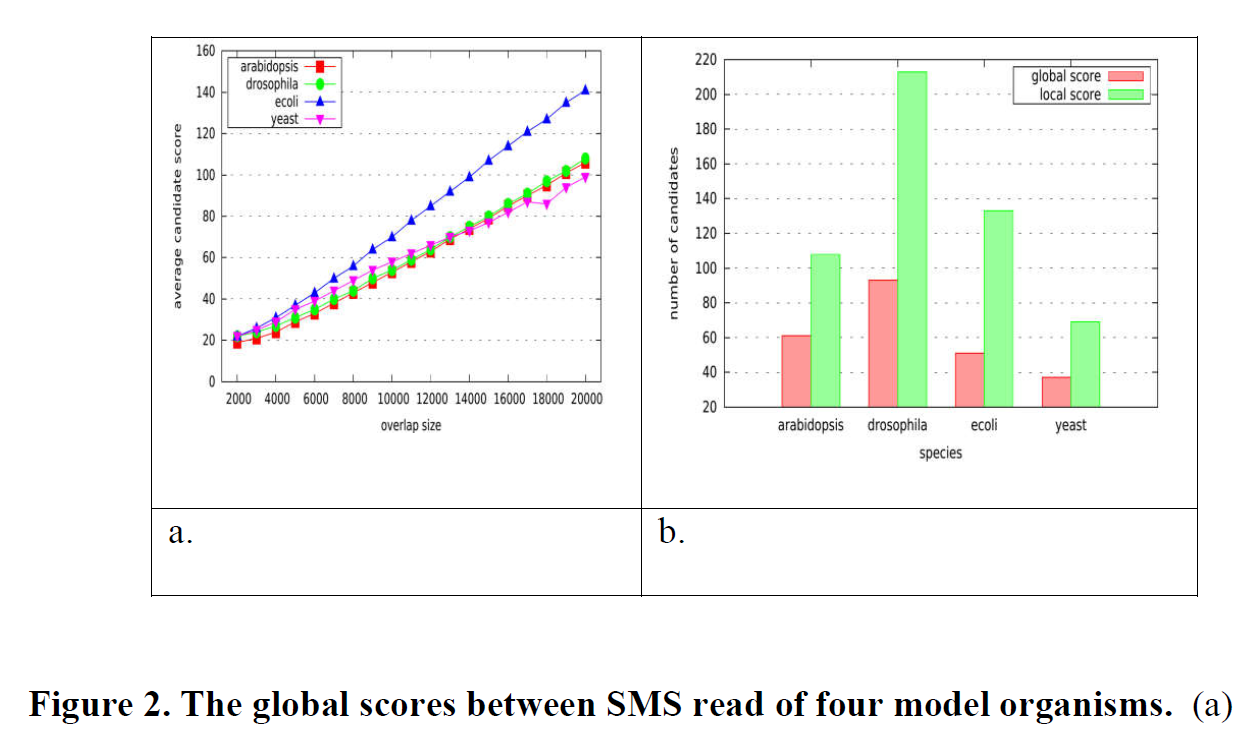
之后选取4个模式生物数据(E coli, Yeast, A.Thaliana and D. Melanogaster)进行测试得到两个结果:a)reads越长相对应得分越高;b)这种打分模式会快节省2~3倍的时间。
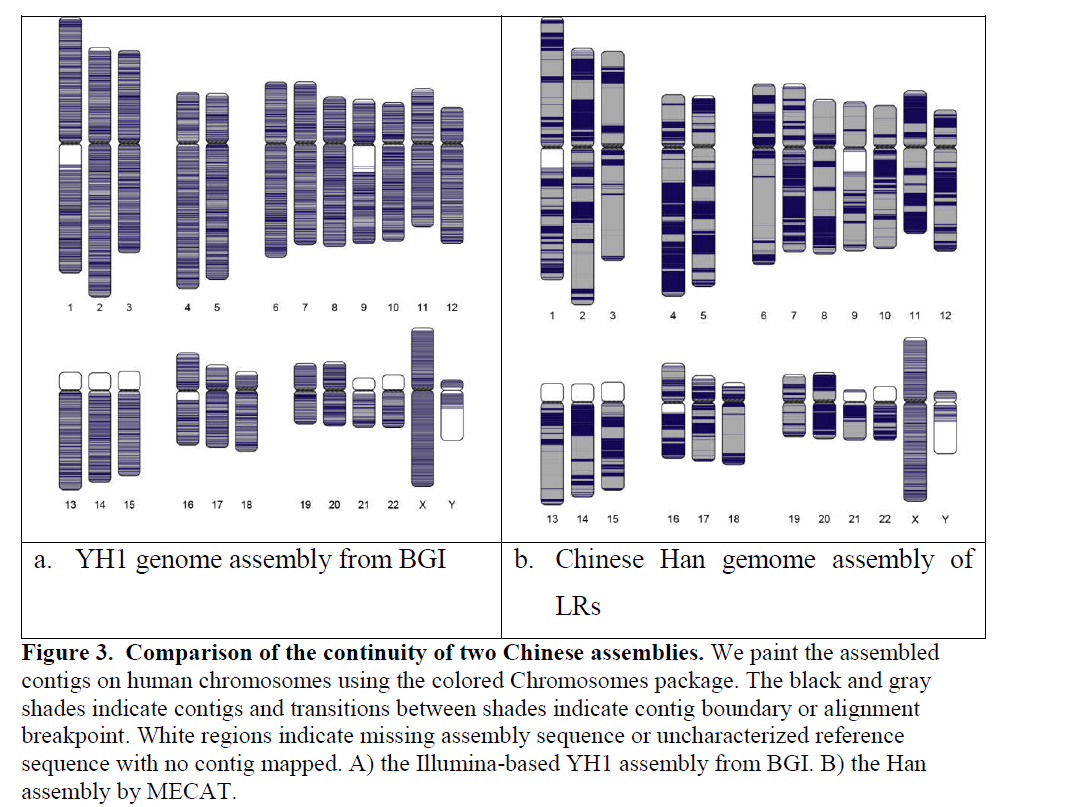
接下来利用真实数据,与其他方法进行对比,结果敏感性和准确性进一步说明MECAT在比对、纠错和组装的综合水平均要优于其他方法。如下面的两个表说明消耗资源、时间、准确行一结合,MECAT软件效果是最好的。
乡亲们,注意哈,MECAT比对,是用的4 Gb不到的内存哦。
貌似哥的手机也可以跑一套~~笑哭。
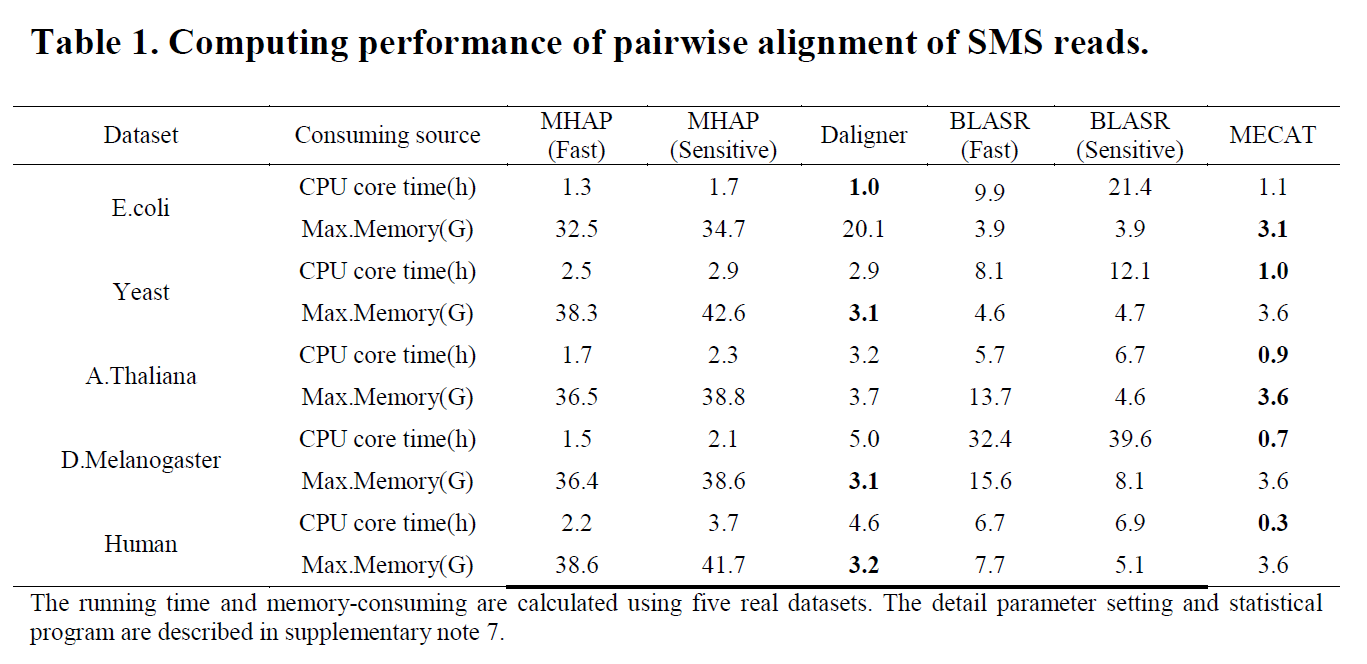
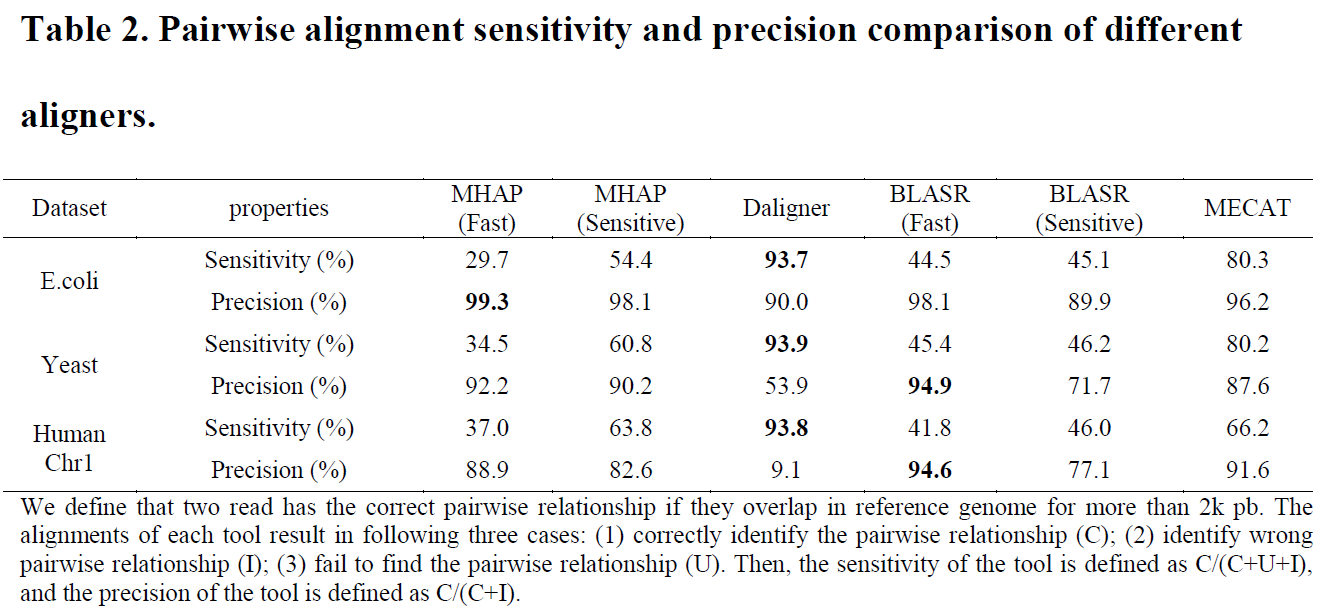
看完这个表,想起京东来了
多快好省
- 发表于 2017-06-26 16:31
- 阅读 ( 9863 )
- 分类:基因组学