文献解读(6)一种用于系统计算诊断和预后基因特征的方法研究
- 荟萃分析
- 功能基因表达分析
- ML方法
- “sigident”
- Sigident DEG,Sigident Enrichment,Sigident Diagnostic和Sigident Prognostic

标题:A Toolbox for Functional Analysis and the Systematic Identification of Diagnostic and Prognostic Gene Expression Signatures Combining Meta-Analysis and Machine Learning
杂志:Cancers IF:6.162 日期:2019.11.15

生物标记物的组合能够代表生物样本中包含的信息,从而支持临床决策。大量研究表明,从微阵列分析中得出的诊断性和预后性基因表达特征具有临床意义。然而,可靠的临床特征受数据集可用性的限制。通过使用数据集合并(meta-nalysis)组合不同的大型队列来人为地增加样本数量是一种有益的解决方案,但是生物标记物识别的方法目前受到限制。有很多R包和在线工具可以进行生物标志物的分析。但是,这些工具专注于特定的疾病和特征类型,仅允许在线分析,而不能根据内部数据计算特征。这些原因限制了它们作为独立工具的使用,从而提出了新的生物信息学方法。机器学习(ML)方法已被证明在医学上很有用。因此,作者开发了一种用于系统计算诊断和预后基因特征的方法,该工具将(1)荟萃分析与(2)功能基因表达分析和(3)ML方法相结合。
今天小编带来的这篇文章【标题:A Toolbox for Functional Analysis and the Systematic Identification of Diagnostic and Prognostic Gene Expression Signatures Combining Meta-Analysis and Machine Learning 杂志:Cancers IF:6.162 日期:2019.11.15】该研究的目标是开发一种具有高预测性能的功能分析和特征计算的通用框架,该框架不限于特定疾病类型,因此可以被广泛的使用。

工具箱的工作流程:作者开发了一个R包“sigident”,该包提供了四个主要功能Sigident DEG,Sigident Enrichment,Sigident Diagnostic和Sigident Prognostic(图1)。
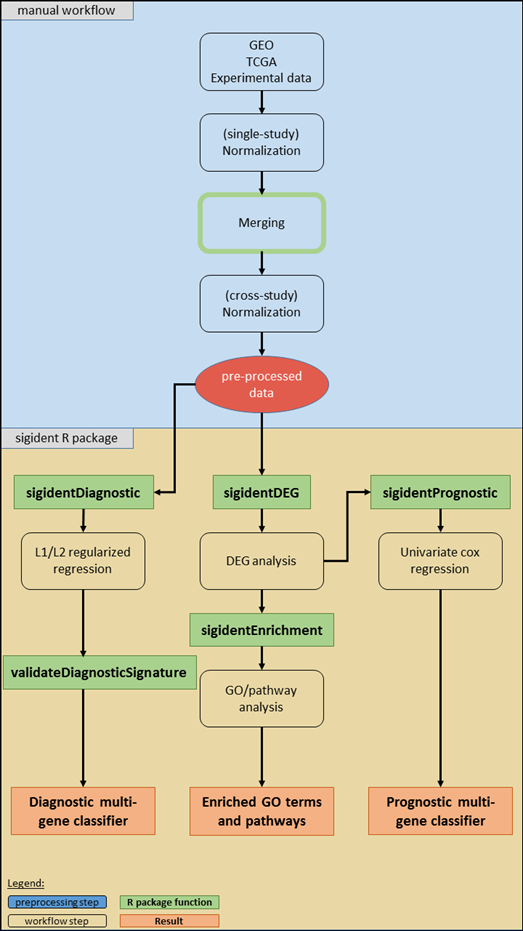 图1.流程图
图1.流程图
作为一个应用示例,作者使用了GEO数据库中三个肺癌数据集(GSE18842,GSE19804和GSE19188)。在把数据合并后将数据集分为训练集(80%)和测试集(20%)来计算诊断signature。此外,作者在三个独立的数据集中(GSE30219,GSE102287,GSE33356)验证了诊断signature。对于预后signature,作者使用GSE19188进行了生存分析,并在两个独立的数据集(GSE30219,GSE50081)中验证了。作者使用了基于inSilicoMerging软件包的代码并结合了limma包进行meta分析(数据集下载,标准化,合并)和功能基因表达分析(DEG分析,热图分析),使用limma包中的goana和kegga函数对DEG进行了GO和KEGG富集分析,使用R包glmnet中的LASSO方法进行诊断signature的分析,使用survival R包对预后signature进行生存利率和风险评分的评估。

1. meta分析(数据集下载,规范化,合并,批效应校正):作者通过分析从GEO数据库下载的三个肺癌数据集来演示该工具箱的工作流程。这包括GSE18842(45个非肿瘤样本,46个肿瘤样本),GSE19804(60个肿瘤, 60个非肿瘤样本)和GSE19188(91个肿瘤,65个非肿瘤样本)。将文件导入R,使用gcrma包进行标准化。合并的数据集包括54,675个转录本和367个样本(197个肿瘤,170个非肿瘤样本)并进行了批次效应的矫正。
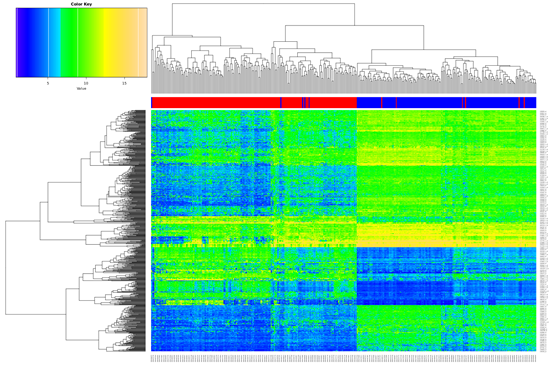
2. 功能基因表达分析:差异表达分析产生了699个在肿瘤和非肿瘤样本中显著差异的基因(图2)。
作者进一步对699个DEG进行了GO和KEGG分析并对特定的通路进行了可视化(图3)。

图3.GO和KEGG通路富集分析
3.诊断和预后signature的计算:接下来,作者分析了合并的数据集(54,675个转录本)以获取诊断signature。首先将合并的数据集分为训练数据集(80%;294个样本)和测试数据集(20%;73个样本)。作者使用L1 / L2正则逻辑回归来拟合广义线性模型来筛选signature。10倍交叉验证得出的λ为0.009260和0.059521(图4,α= 1)。图4显示了用于预测测试数据样本的LASSO signature的交叉验证误差(左)和混淆矩阵(右)。
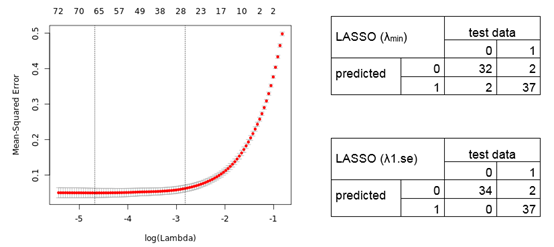
图4. 根据训练肺癌数据集上的lambda的对数进行10倍交叉验证的均方误差
作者进一步应用了弹性网络回归。弹性网络模型计算的预测测试数据样本的交叉验证误差(左)和结果混淆矩阵(右)如图5所示。
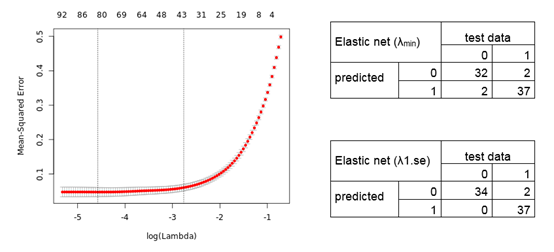 图5.弹性网络回归模型
图5.弹性网络回归模型
接下来就是验证部分,作者在三个独立的数据集中验证了基因signature(GSE30219,293个肺癌, 14个非肺癌样本; GSE102287,32个肺癌, 34个非肺癌样本; GSE33356,60个肺癌,60个非肺癌样本; 54,675个基因)。验证的结果显示在图6中,结果表明该方法对肺癌和非肺癌样本的分类具有高诊断能力。
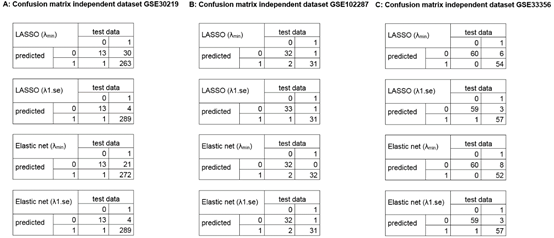 图6. 独立数据集中已识别诊断signature的混淆矩阵
图6. 独立数据集中已识别诊断signature的混淆矩阵
在确定诊断signature后,作者测试了相关的预后signature。因此作者使用单变量Cox风险回归模型分析了699个DEG对患者生存的影响。Cox回归分析显示22个DEG对患者存活率有显著影响。
接下来,作者使用一种算法训练了预后22基因分类器,该算法比较了合并数据集GSE18842和GSE19804的肿瘤样本与健康样本之间的表达谱。在这里,作者测试了22个基因标记是否可以对高和低死亡风险的患者进行分类。因此,作者使用训练的分类器将患者样本分为高风险和低风险组。结果显示这22个基因标记可以对高风险和低风险患者进行分类(图7)。这表明已鉴定的22个基因分类器反映了可以区分高风险和低风险肿瘤疾病的主要肿瘤因素的常见预后特征。
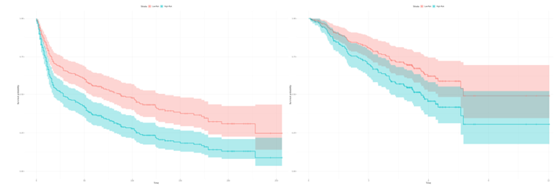 图7.Kaplan-Meier分析
图7.Kaplan-Meier分析

该研究开发了一种用于诊断和预后基因signature识别的有效工具箱。它是第一个将meta分析与基因表达分析和ML方法相结合的R软件包工具,用于系统地计算统计上可靠的基因signature。重要的是,该工具不限于特定疾病。该工具为有效分析数据和更好地对疾病进行临床管理打开了新的窗口。

1. Borrebaeck, C.A.K. Precision diagnostics: Moving towards protein biomarker signatures of clinical utility incancer. Nat. Rev. Cancer 2017, 17, 199–204.
2. Kunz, M.; Wolf, B.; Schulze, H.; Atlan, D.; Walles, T.; Walles, H.; Dandekar, T. Non-coding rnas in lung cancer: Contribution of bioinformatics analysis to the development of non-invasive diagnostic tools. Genes 2016 , 8, 8.
3. Aguirre-Gamboa, R.; Trevino, V. Survmicro: Assessment of miRNA-based prognostic signatures for cancer clinical outcomes by multivariate survival analysis. Bioinformatics 2014 , 30, 1630–1632.
4. Cusumano, P.G.; Generali, D.; Ciruelos, E.; Manso, L.; Ghanem, I.; Lifrange, E.; Jerusalem, G.; Klaase, J.;de Snoo, F.; Stork-Sloots, L.; et al. European inter-institutional impact study of mammaprint. Breast 2014 , 23, 423–428.
5.Montani,F.;Marzi,M.J.;Dezi,F.;Dama,E.;Carletti,R.M.;Bonizzi,G.;Bertolotti,R.;Bellomi,M.;Rampinelli,C.;Maisonneuve, P.; et al. miR-test: A blood test for lung cancer early detection. J. Natl. Cancer Inst. 2015 , 107.

文献解释(1)什么?这篇文章为预测预后和发现抗转移药物提供了帮助?
- 发表于 2019-12-30 17:49
- 阅读 ( 4421 )
- 分类:文献解读
