干货|基因注释方法论
简单介绍基因预测方法
现如今随着测序技术的发展,组装一个完整的基因组也是越来越普遍,价格已经不像前些年那样昂贵(当然超大基因组除外),待基因组组装完成后,基因预测将会是接下来需要进行的一个重要工作(虽然能编码的基因相对于整个基因组只是占了很小一部分),但通过这些基因结合一些近缘物种,还是能挖掘一些比较有意思东西,如物种进化等。那么接下来小编就给简单介绍一些基因预测方面方法。
目前,基因组预测策略大致可以分为三种:Ab initio、Homology-based和EST/Unigene。
从头预测
Ab initio主要通过探索DNA序列中特异的区域,如基因的起始区域和终止区域,来进行基因预测。目前常用的软件有Augustus、GlimmerHMM、SNAP、GeneID、GenScan、Brak等。
Augustus运用隐马尔科夫模型,模型在DNA序列和基因结构上定义一个概率分布,采用维特比的算法,它自身带了一个训练集,如人、斑马鱼等。在进行预测是可以选择自带的训练集,也可以用挑选转录组和同源预测最优结果给它生成一个训练集。这里顺带说下Braker软件,它是基于genemaker预测结果作为训练集,通常小编Augustus和Braker会二选一。
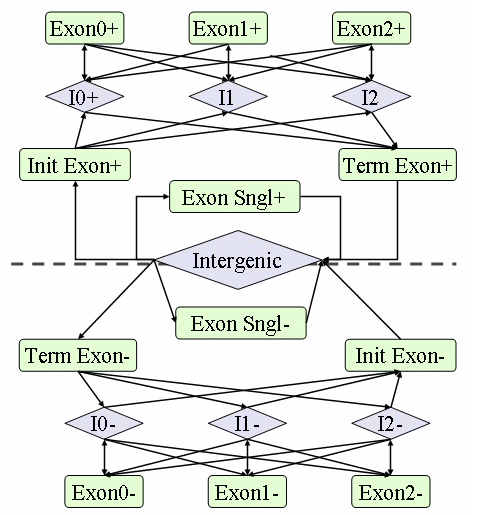
GlimmerHMM是把一个基因看做几种特征序列,这些特征序列包括内含子、基因间区和四种外显子(初始、中间、最终和单一外显子)之后进行有序切换形成马尔科夫链。示意图如下:
GlimmerHMM使用的模型基于以下几个假设:
- 假设每个基因都开始于起始密码子ATG
- 假设每个基因阅读框内除最后一个密码子外没有终止密码子(no in-frame stop codons)。
- 每个外显子与前一个外显子在同一个阅读框中。(翻译阅读时外显子间没有移框).
它也是需要一个训练集,通常也是自己生成一个训练集的效果会略优于已有的一些。(http://ccb.jhu.edu/software/glimmerhmm/man.shtml)
SNAP通过隐马尔科夫模型进行预测,也是需要一个训练集。
以上这些软件都可以自身构建一个训练集,这里小编觉得毕竟还是用自己的东西舒服,也就是自身训练结果要稍微优于其他模式生物训练集。这里小编在做真菌时,从头软件一般会选取这三个,GenScan和GeneID就放弃掉了,动植物基因组通常就是多多益善吗,能用上就都给用上。
GenScan也是一款比较经典软件,通常在预测真核生物(人)还是有不错的效果。
GeneID可以算是元老级,第一代的基因识别软件,这个准确率不高,通常在整合是权重也不会给太高。
同源预测
同源预测软件通常利用GeneWise和GeneMoMa,前者是需要同源物种的蛋白序列,后者需要同源物种基因组序列及对应的GFF文件,目前小编已经抛弃GeneWise,使用最多的就是GeneMoMa,但是让小编十分头疼的是在准备GFF文件太花费精力,这个软件真的是挑肥拣瘦,必须满足其格式才能可以运行,目前从NCBI的Reseq和Ensemble上下载都可以,其他地方来的那就得还点时间写个脚本改下了。
转录组数据预测
PASA软件是基于Unigene/EST序列进行预测软件,这个可能就需要拿到一个混样转录组数据首先进行无参组装,接下来根据Unigene组装结果在进行比对,通常用Gmap或Blat两种方法,最好三代全长转录本和二代一起来进行预测,这样可以使得找到的结构更为准确、可靠,此外PASA还有另外的一个功能就是可以用其预测可变剪切,俗称PASA修饰。
最终结果整合
这么多软件跑出来的结果,有的可靠性高些,比如转录组和同源;有一些要稍微差一些如GeneID,那么就需要一个软件将这些结果进行一个整合,通俗些就是大家放到一起比较下,看下各个软件预测结果分布情况,本着以少数服从多数原则(这里只是简单比喻下莫要当真),根据权重打分,使用EVM软件得到一版最终结果,目前小编用到最多的就是EVM真菌、植物或动物统统可以搞定,用过一段时间Glean,感觉在整合超大基因时,容易成多个(或许是参数没有调整合理)。
总结:
上面就是小编在进行基因预测时的一些软件使用心得,还有是再做一些研究比较多的物种比如水稻等,那同源权重一定要调高,毕竟人家预测出来的经过验证的,在我们的结果中理论上是应该存在,这样才能说明我们预测结果是靠谱的嘛;如果研究不是很多,同源比较少,那可以适当调高从头软件权重,主要应该以转录组为核心。
- 发表于 2017-08-02 20:36
- 阅读 ( 9423 )
- 分类:基因组学