如何极其简单的使用GEO数据来做差异分析
新版本DECenter使用看这里:https://www.shengxin.ren/article/207
无论你是要看某个基因是否差异表达或者筛选某个GEO数据集的差异基因,这个方法绝对能够帮助你事半功倍
首先假设你已经找到了一套数据GSE32323
这套数据共包含44个样本,其中有17个配对的癌与癌旁样本
我们先下载数据,如图
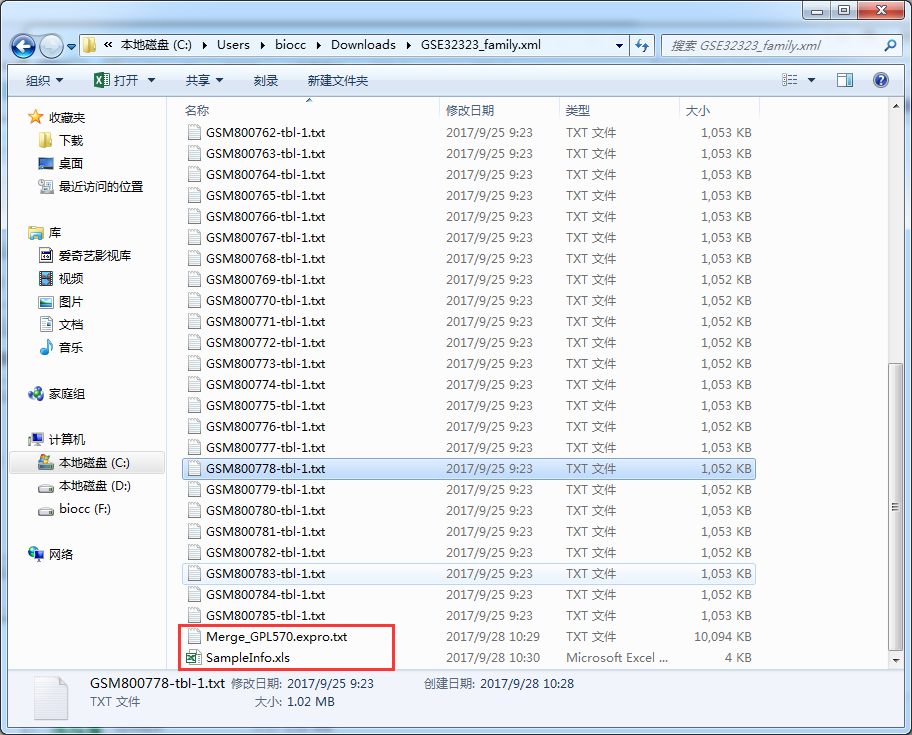
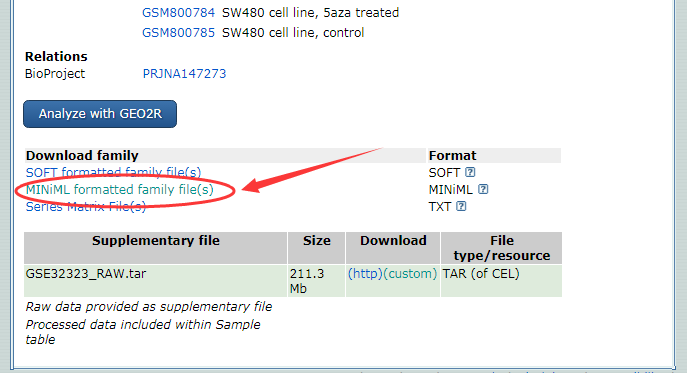 然后使用GEO芯片数据转换器提取出表达矩阵和样本信息表 (不会提看这里)如图:
然后使用GEO芯片数据转换器提取出表达矩阵和样本信息表 (不会提看这里)如图:
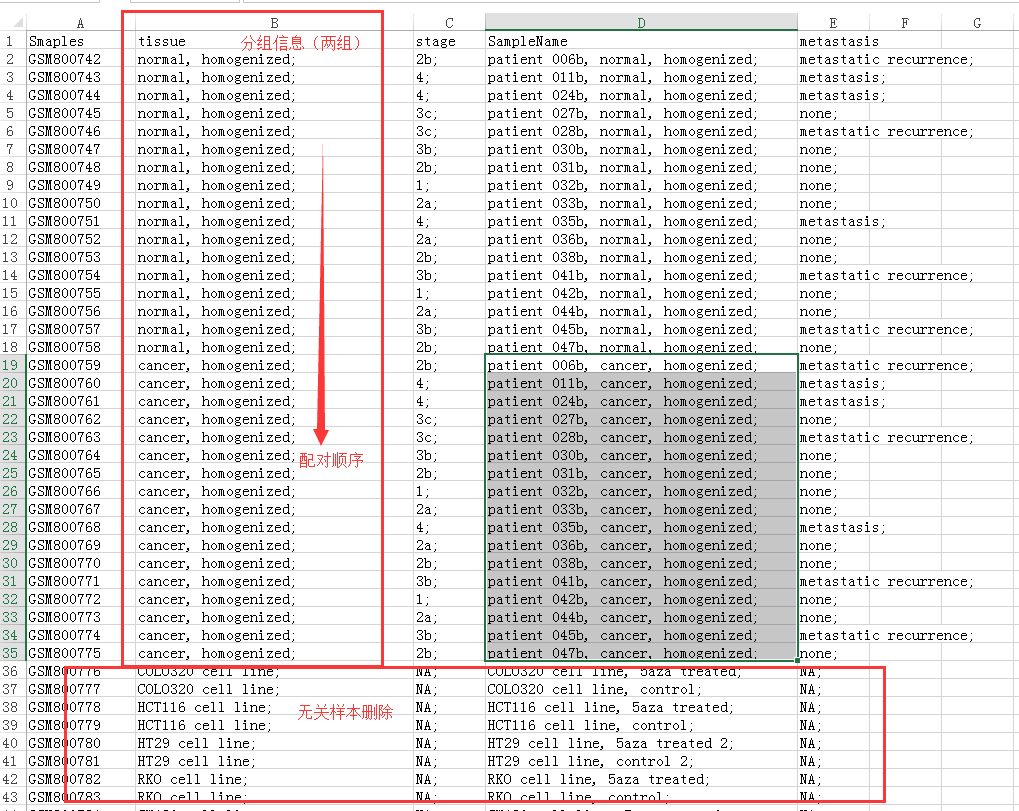
打开SampleInfo.xls文件编辑样本顺序和分组
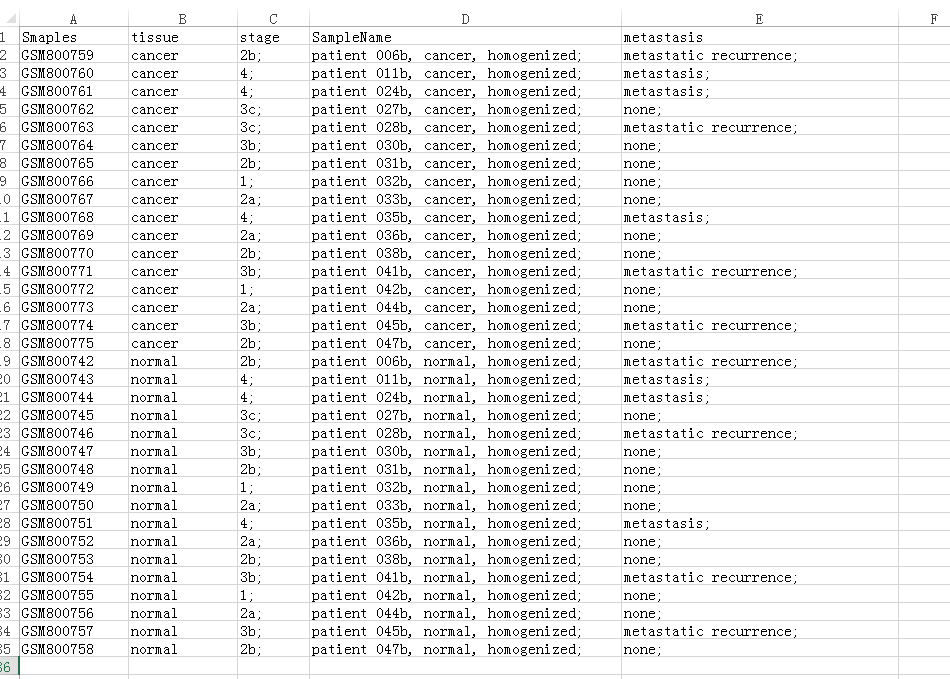
 最终修改后的样本信息表为(为什么cancer排在前面?软件默认前面比后面啊):
最终修改后的样本信息表为(为什么cancer排在前面?软件默认前面比后面啊):
然后使用DECenter(下载链接:http://gap.shengxin.ren/tool/10/)进行差异分析
打开,长这个样子
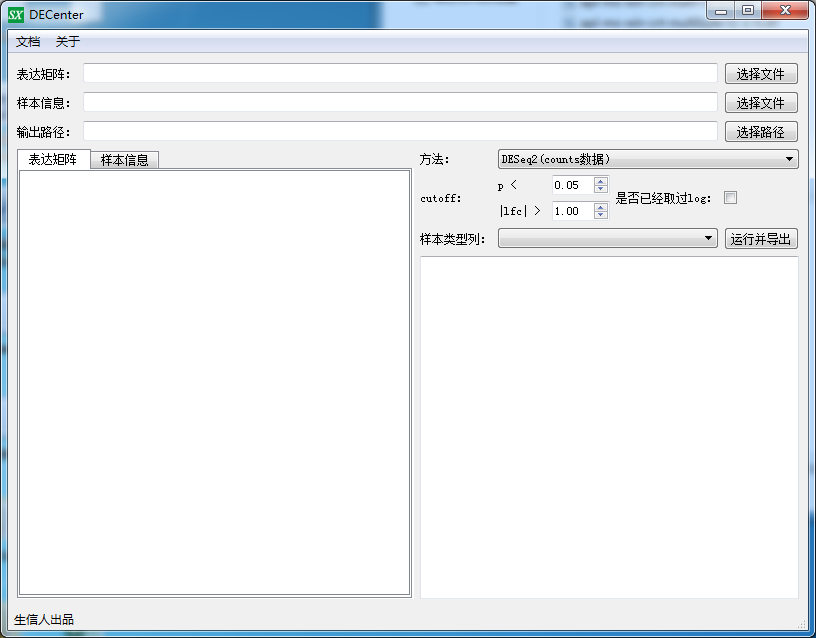
看似需填项比较多,其实无非就四步
1、选择表达矩阵
2、选择样本信息表(因为你要告诉软件怎么分组的嘛)
3、选择筛选差异的方法,如果是芯片数据当然选择limma了,如果是RNA-Seq的counts数据,选择DESeq2或者edgR也行啊
4、选择样本分组列,然后选择结果保存目录点击运行就行了
示例如下:
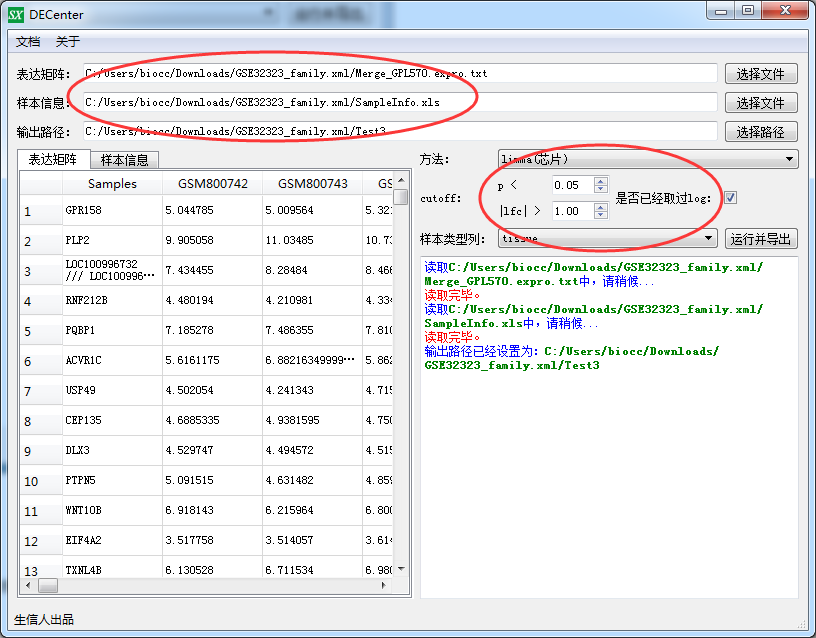 等提示跑完,打开结果保存的目录
等提示跑完,打开结果保存的目录
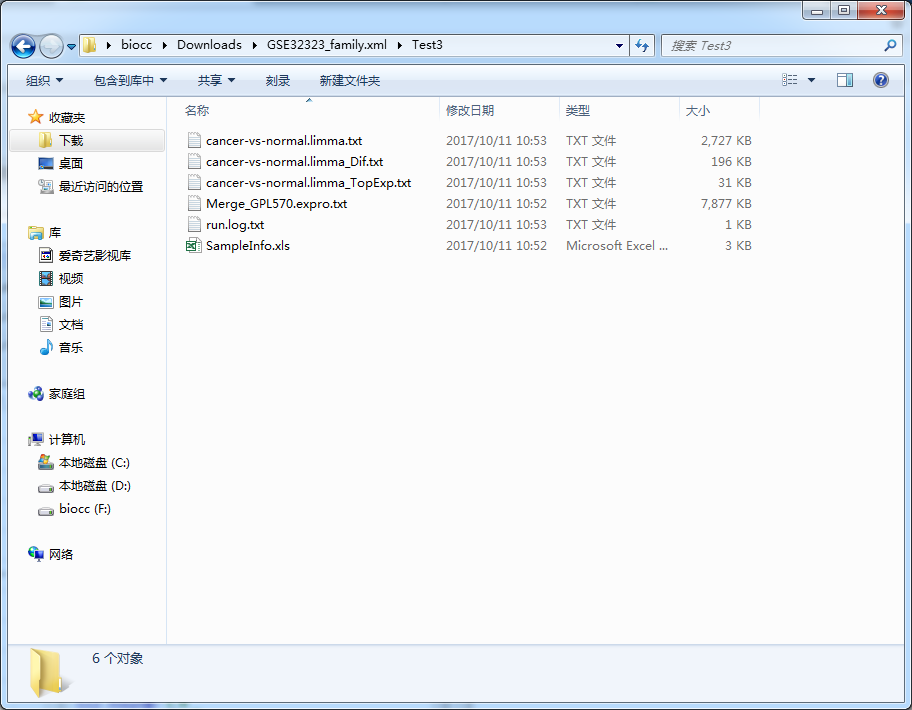 总共得到三个文件
总共得到三个文件
1、cancer-vs-normal.limma.txt:这是cancer比normal的所有基因差异结果
2、cancer-vs-normal.limma_Dif.txt:这是根据咱们在软件选择的阈值筛选之后的差异结果
3、cancer-vs-normal.limma_TopExp.txt:这是提取的最差异的基因的表达谱
得到了差异结果之后,我们要对结果进行一个展示吧
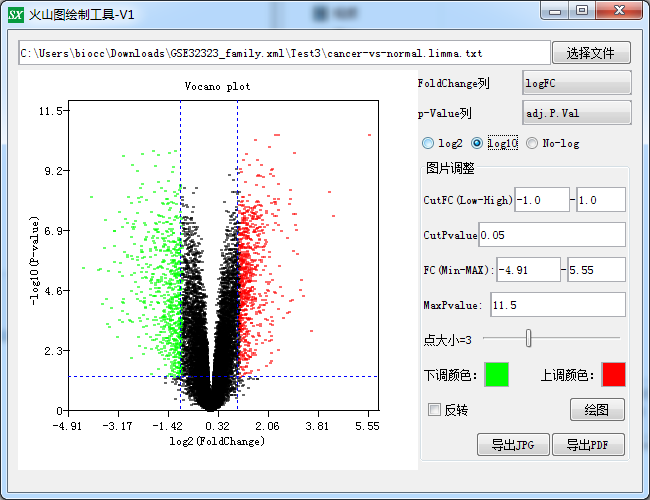
先拿cancer-vs-normal.limma.txt使用火山图制作工具做个火山图,教程看这里:https://www.shengxin.ren/article/174
 再拿cancer-vs-normal.limma_TopExp.txt使用热图绘制工具画个热图:教程看这里:https://www.shengxin.ren/article/165
再拿cancer-vs-normal.limma_TopExp.txt使用热图绘制工具画个热图:教程看这里:https://www.shengxin.ren/article/165
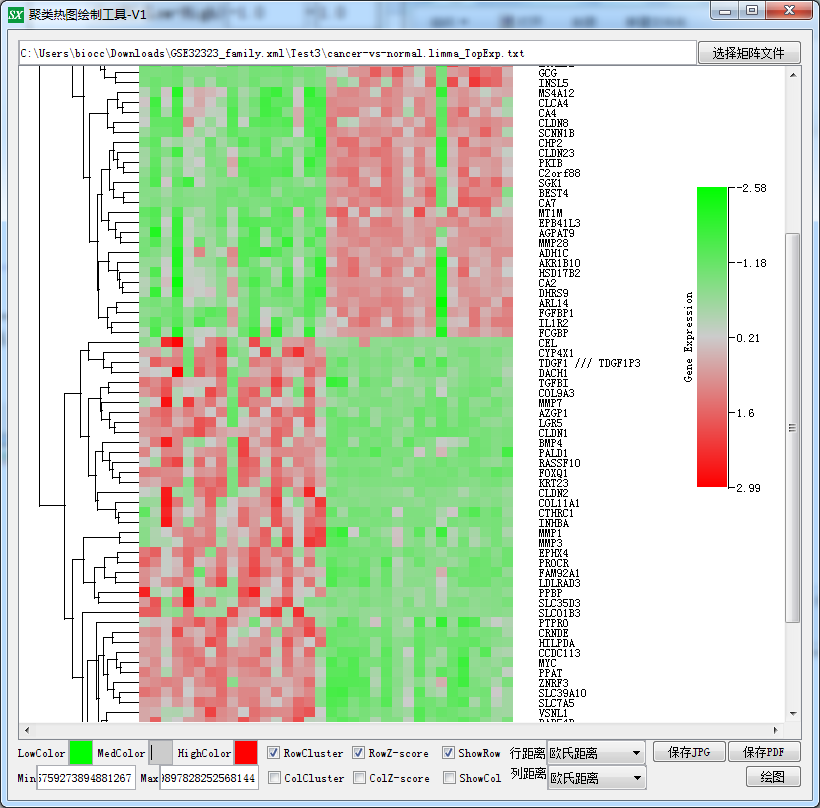 导出图片,放到PPT中就可以向老板汇报了
导出图片,放到PPT中就可以向老板汇报了
- 发表于 2017-09-28 10:48
- 阅读 ( 77173 )
- 分类:软件工具
