limma的两个弟弟:edgeR和DESeq2
01
—
研究背景
上一篇公众号我们为大家详细的介绍了R软件包limma筛选差异基因,limma包做差异分析要求数据满足正态分布或近似正态分布,如基因芯片、TPM格式的高通量测序数据。随着高通量测序价格的降低,RNA-seq测序技术与芯片测序技术相比具有通量高,GC偏好性较小,能发现未知的转录本等特点[1],越来越多的科研人员选择用转录组高通量测序技术来代替传统的芯片测序技术。高通量测序得到的原始数据为fastq文件,经过数据质控,比对,定量之后得到count矩阵。通常认为Count数据不符合正态分布而服从泊松分布。对于count数据来说,用limma包做差异分析,误差较大,所以小编今天给大家介绍另外两个计算差异基因的方法,分别为edgeR[2]和DESeq2[3]。值得一提的是edgeR和limma是由一个团队开发的,算法有点过时了,DESeq2目前使用频率较高。我们平台不但集成了这两个主流的分析方法,同时对差异分析结果进行可视化,只需输入表达矩阵和分组信息,点击鼠标就可完成整个差异分析,老板再也不用催我敲代码了。
02
—
使用方法
1.输入网址:http://sangerbox.com/Tool
点击“转录组Count数据差异分析工具”即可进入分析界面
2.输入数据格式 表达矩阵:行名为ENSG ID,这里小编根据团队的项目经验说明一下,这里使用的是ENSG ID ,并没有将其转化为Gene Symbol。原因是由于将ENSG ID转化为Gene Symbol,如果将一个Gene Symbol对应多个ENSG ID去中值或者取均值,得到的新矩阵会含有小数值。而edgeR和DEseq2这两个软件要求输入的文件中所有数值必须要是整数,所以小编以前在做项目的过程中先用ENSG ID做差异分析,然后得到差异分析结果以后,在将ENSG ID转化为Gene Symbol。列名为样本名称,下图所示。 
分组矩阵:共两列,第一列为样本名称,要与表达矩阵的样本名一一对应,第二列为样本的分组信息,如normal与tumour如下图所示
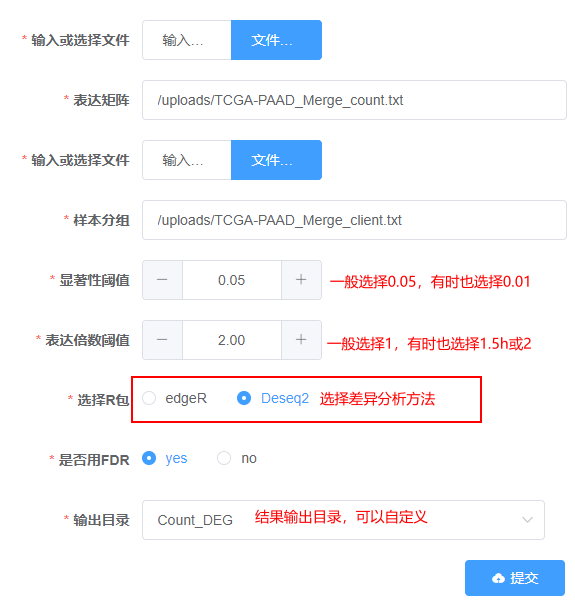
3.参数设置
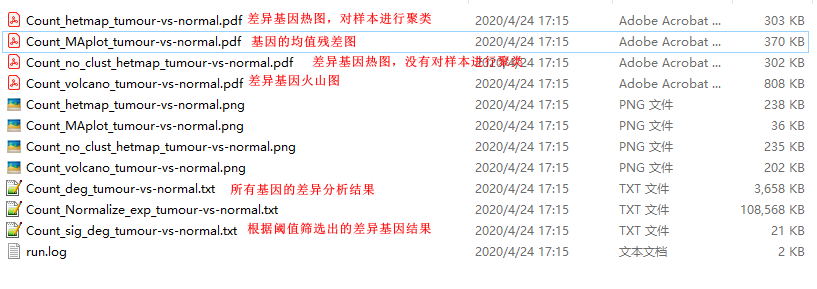
4.结果目录:
在个人中心有结果目录,如果事先不指定运行结果目录,默认输入到Count_DEG目录下,结果如下图所示:
参考文献
[1] Agarwal A, Koppstein D, Rozowsky J, et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 2010;11:383. Published 2010 Jun 17.[2] Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140. [3] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
具体指引详见:

- 发表于 2020-05-26 11:09
- 阅读 ( 8383 )
- 分类:软件工具
