使用TCGA做CeRNA又有了便捷的方法
在早先时候我介绍了CeRNA的一些做法,尤其是怎么构建CeRNA网络和怎么去对网络做剪枝,在视频中都是针对于不需要编程去操作,实际上如果你会R的话,下面介绍的工具会让你事半功倍。
这是一篇上...
在早先时候我介绍了CeRNA的一些做法,尤其是怎么构建CeRNA网络和怎么去对网络做剪枝,在视频中都是针对于不需要编程去操作,实际上如果你会R的话,下面介绍的工具会让你事半功倍。
这是一篇上周发表在Bioinfomatics上的文章:
这边文章主要是建立了这样的一个workflow来构建CeRNA及后续的分析,以TCGA的胆管癌为例:TCGA-CHOL
1、首先 安装R包
source("https://bioconductor.org/biocLite.R")
biocLite("GDCRNATools")
2、下载数据,作者使用了GDC API 自动下载TCGA的数据,命令如下
library(GDCRNATools)
project <- 'TCGA-CHOL'
rnadir <-paste(project, 'RNAseq', sep='/')#设置RNASeq文件保存目录
mirdir <-paste(project, 'miRNAs', sep='/')#设置miRNA文件保存目录
####### Download RNAseq data #######
gdcRNADownload(project.id ='TCGA-CHOL',data.type='RNAseq',write.manifest =FALSE,directory= rnadir)
####### Download miRNAs data #######
gdcRNADownload(project.id='TCGA-CHOL',data.type='miRNAs',write.manifest =FALSE,directory= mirdir)
3、数据解析和预处理
####### Parse RNAseq metadata #######
metaMatrix.RNA <-gdcParseMetadata(project.id ='TCGA-CHOL',data.type ='RNAseq',write.meta =FALSE)
####### Filter duplicated samples in RNAseq metadata #######
metaMatrix.RNA <-gdcFilterDuplicate(metaMatrix.RNA)
####### Filter non-Primary Tumor and non-Solid Tissue Normal samples in RNAseq metadata #######
metaMatrix.RNA <-gdcFilterSampleType(metaMatrix.RNA)
####### Parse miRNAs metadata #######
metaMatrix.MIR <-gdcParseMetadata(project.id ='TCGA-CHOL',data.type ='miRNAs',write.meta =FALSE)
####### Filter duplicated samples in miRNAs metadata #######
metaMatrix.MIR <-gdcFilterDuplicate(metaMatrix.MIR)
####### Filter non-Primary Tumor and non-Solid Tissue Normal samples in miRNAs metadata #######
metaMatrix.MIR <-gdcFilterSampleType(metaMatrix.MIR)
4、合并表达矩阵,得到成熟体miRNA的表达谱和mRNA的表达谱,数据使用的是Count数据
####### Merge RNAseq data #######
rnaCounts <-gdcRNAMerge(metadata = metaMatrix.RNA, path = rnadir, data.type ='RNAseq')
####### Merge miRNAs data #######
mirCounts <-gdcRNAMerge(metadata = metaMatrix.MIR,path= mirdir,data.type ='miRNAs')
5、标准化Counts数据,使用的是edgR 转TMM再使用limma的voom函数进行标准化
####### Normalization of RNAseq data #######
rnaExpr <-gdcVoomNormalization(counts = rnaCounts, filter =FALSE)#filter =TRUE则去除logcpm < 1样本占比大于一半的基因
####### Normalization of miRNAs data #######
mirExpr <-gdcVoomNormalization(counts = mirCounts, filter =FALSE)
6、差异基因筛选,作者这里提供了多种方法筛选差异基因,limma|edgR|DEseq2
DEGAll <-gdcDEAnalysis(counts= rnaCounts, #Counts数据
group=metaMatrix.RNA$sample_type, #样本分组信息
comparison ='PrimaryTumor-SolidTissueNormal',#比较组信息
method='limma')
### All DEGs
deALL <- gdcDEReport(deg = DEGAll, gene.type = 'all')
### DE long-noncoding
deLNC <- gdcDEReport(deg = DEGAll, gene.type = 'long_non_coding')
### DE protein coding genes
dePC <- gdcDEReport(deg = DEGAll, gene.type = 'protein_coding')
7、开始构建CeRNA网络,作者这里考虑了mRNA和lncRNA共享的miRNA的个数构建了超几何分布模型来评估mRNA-lncRNA互作的可能性,同时考虑了表达的相关性
ceOutput <- gdcCEAnalysis(lnc = rownames(deLNC),
pc = rownames(dePC),
lnc.targets = 'starBase', #选择lncRNA-miRNA互作数据库
pc.targets = 'starBase', #选择mRNA-miRNA互作数据库
rna.expr = rnaExpr,
mir.expr = mirExpr)
#######自定义互作数据lncTarget、pcTarget######
ceOutput <- gdcCEAnalysis(lnc = rownames(deLNC),
pc = rownames(dePC),
lnc.targets = lncTarget,
pc.targets = pcTarget,
rna.expr = rnaExpr,
mir.expr = mirExpr)
8、导出CeRNA网络
ceOutput2 <- ceOutput[ceOutput$hyperPValue<0.01 &
ceOutput$corPValue<0.01 & ceOutput$regSim != 0,]
edges <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'edges')
nodes <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'nodes')
write.table(edges, file='edges.txt', sep='\t', quote=F)
write.table(nodes, file='nodes.txt', sep='\t', quote=F)
9、到此CeRNA网络构建完成
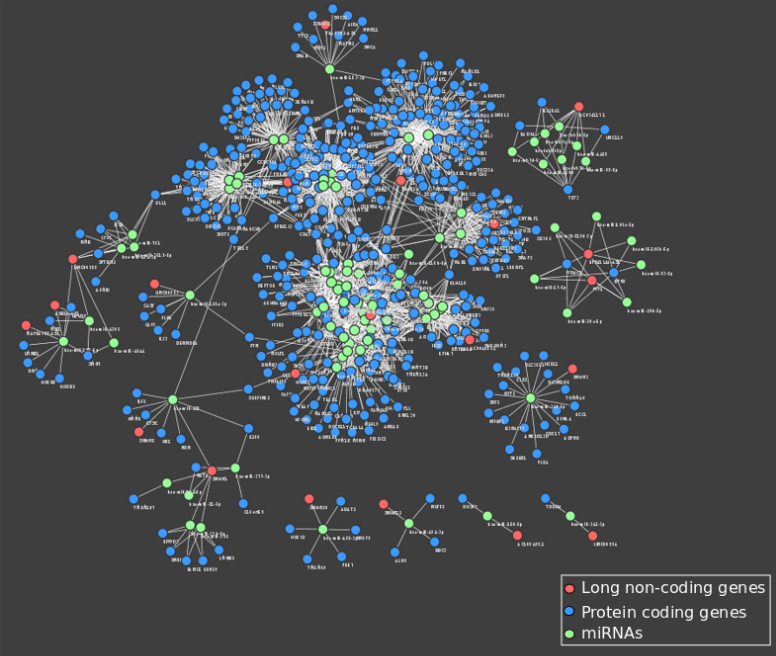
更多详情请参考:http://bioconductor.org/packages/devel/bioc/vignettes/GDCRNATools/inst/doc/GDCRNATools.html#case
- 发表于 2018-08-13 09:52
- 阅读 ( 10132 )
- 分类:软件工具
