两篇胃癌lncRNA signature文章解读之二(IF=3.2)
这是一篇2017年的文章,文章从lncRNA的角度筛选lncRNA signature,该文章发表在Journal of Cancer上,影响因子如下:
 文章题目为:Differentially Expressed lncRNAs in Gastric Cancer Patients: A Potential Biomarker for Gastric Cancer Prognosis
文章题目为:Differentially Expressed lncRNAs in Gastric Cancer Patients: A Potential Biomarker for Gastric Cancer Prognosis

还是胃癌,还是lncRNA signature,与之前解析的那篇七分多的做的是一样的事情。
我们简单的来看一下摘要
 从中大体上可以看出作者首先筛选了差异的lncRNA,然后使用lasso进行降维得到12个lncRNA,最后使用这12个lncRNA构建预后模型对样本进行高低风险分类,功能分析显示高风险样本与癌症的药物治疗和转移相关。
从中大体上可以看出作者首先筛选了差异的lncRNA,然后使用lasso进行降维得到12个lncRNA,最后使用这12个lncRNA构建预后模型对样本进行高低风险分类,功能分析显示高风险样本与癌症的药物治疗和转移相关。
大体上思路与之前的那篇文章出奇一致,但是方法不同。
我们来一步一步解析一下这篇文章
1、数据来源及下载
作者从GEO数据库中选择了三套数据集:GSE79973、GSE62254、GSE15459,其中GSE79973数据集包含了10对配对的癌与癌旁样本,GSE62254、GSE15459两套数据即在上一篇文章中使用过,作者下载了这三套数据集的原始数据CEL,然后去除了没有随访信息的样本,使用GSE62254作为建模的训练集,使用GSE15459作为建模的验证集。
2、数据的处理
3、差异分析
我们使用R包limma对10对癌与癌旁的样本的lncRNA表达谱进行差异分析,选择p<0.01作为阈值(这里没有考虑foldchange哦),最终得到了339个差异的lncRNA.
4、lasso分析降维
我们使用这339个差异的lncRNA,从训练集数据中提取这些lncRNA表达谱,结合随访信息,使用lasso cox回归进行降维,最终得到12个lncRNA。
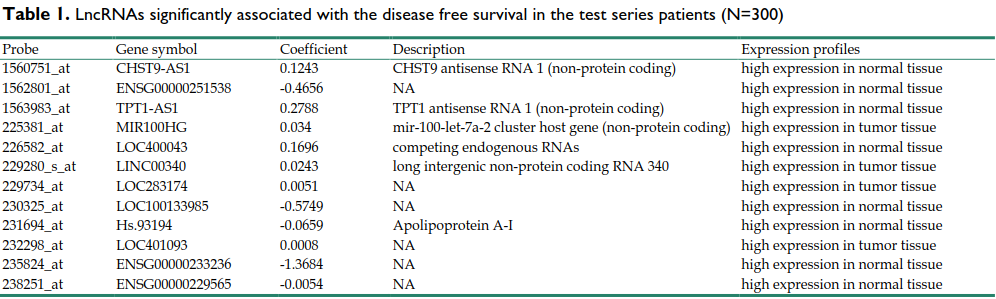
5、建立预后模型
使用lasso cox的回归系数作为模型的系数(表格中第三列),建立预后模型公式:
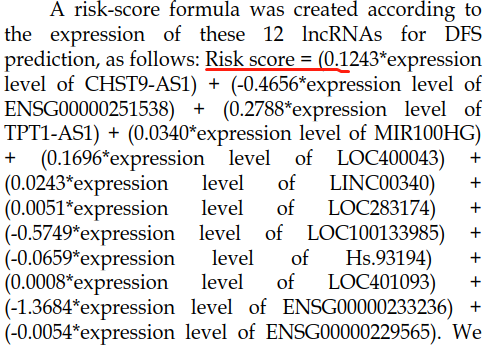
6、根据模型计算每个样本的风险得分
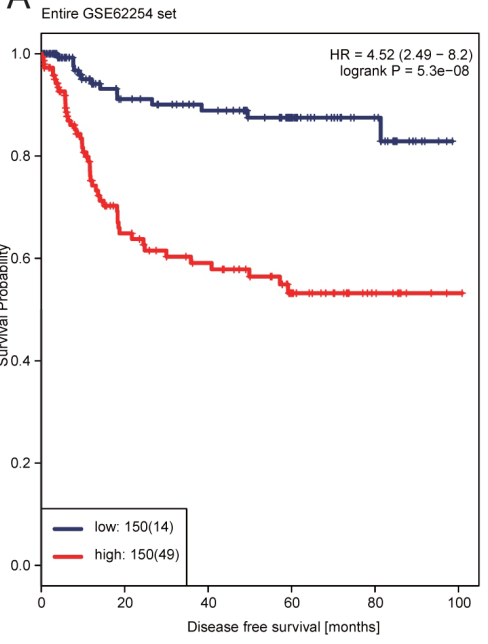
模型公式已经得到了,这个时候我们只需要将每个样本中对应的那12个lncRNA的表达水平提取出来,然后带入这个公式就可以得到这个样本的风险得分了,这个时候我们共有300个样本就得到了300个样本的风险得分,我们根据这得分的高低对样本进行平均分类,分成得分高的一组150个,得分低的一组、150个,然后再进一步分析两组的预后差异,发现他们具有显著的预后差异如图:
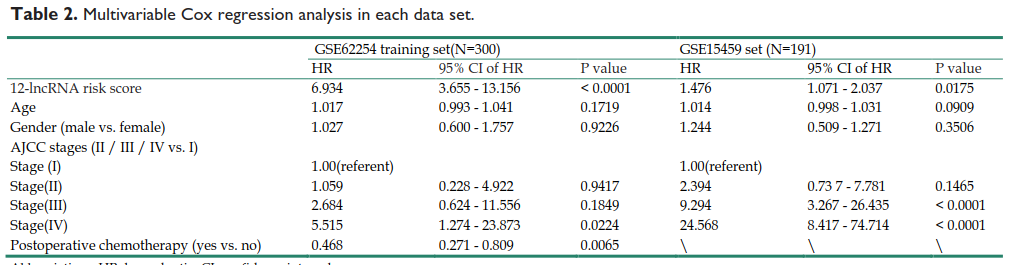
 进一步的我们比较了这些风险得分的预后价值与临床其他特征(TNM)的预后价值,如图,可以看出这个风险模型的p值极显著,而其他几个临床特征p值很大,这说明这个模型的预后价值比TNM Stage好。
进一步的我们比较了这些风险得分的预后价值与临床其他特征(TNM)的预后价值,如图,可以看出这个风险模型的p值极显著,而其他几个临床特征p值很大,这说明这个模型的预后价值比TNM Stage好。
根据每个样本的风险得分,作者分析了复发组样本和无复发组样本的风险得分的分布如图,红色表示复发样本,从A图中我们可以看出复发组样本的风险得分普遍较高(偏右),从B图可以看出复发组样本的风险得分的分布明显比无复发组的风险得分要高(偏右),这提示了风险得分高的样本更有可能发生复发事件。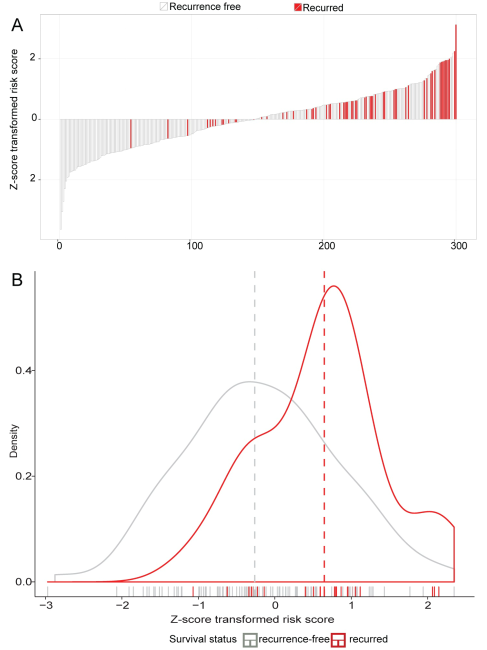
7、测试数据集验证
我们用测试数据集中的这12个lncRNA的表达谱来计算每个样本的风险得分,然后使用中位数来看一下能不能对样本进行高低风险分析如图,发现也能够将样本分开。
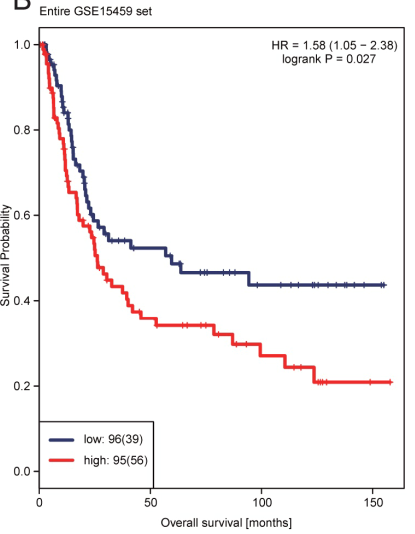 8、模型的适用性探索
8、模型的适用性探索
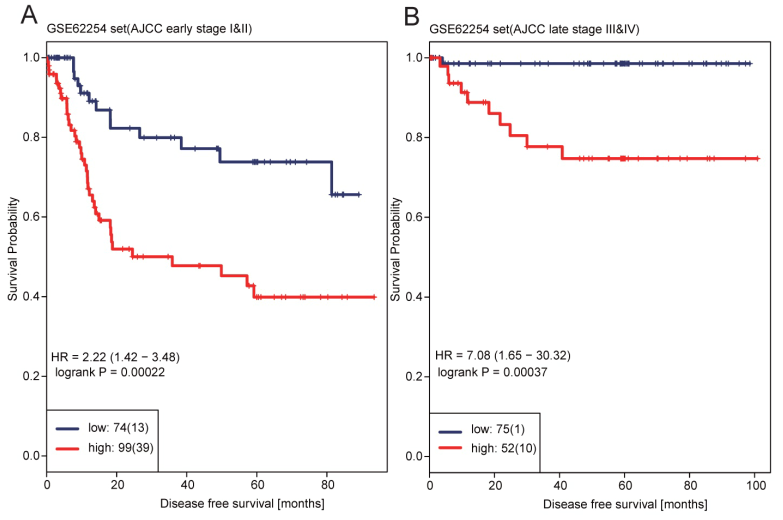
我们使用的是所有样本进行的建模和验证,为了进一步观察模型的适用范围,我们根据样本的Stage分成高Stage和低Stage两组样本,然后分别使用模型去对样本进行分类,看看分类的效果如何,如图,从中可以看出在两个阶段的病人中依然能够对样本进行分类,这提示了模型不仅可以用于早期的筛选,也可以用于晚期的筛选。
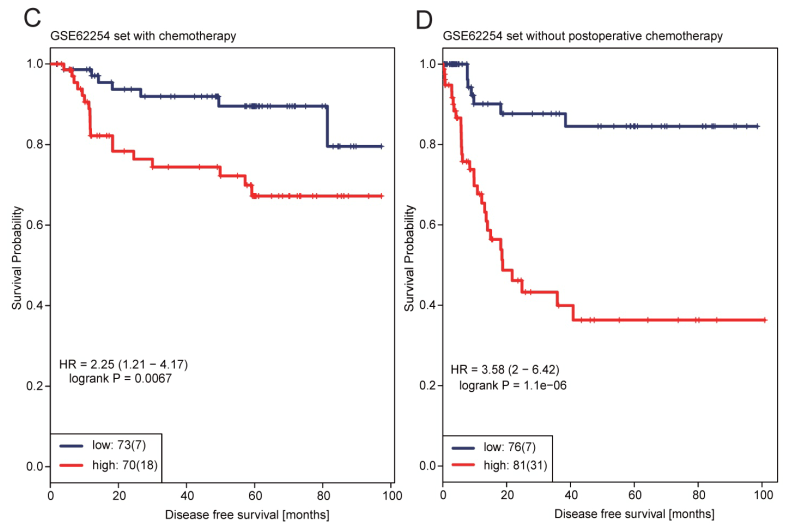
 进一步的,我们将 是否化疗的病人分开来进行分类,看看模型能不能把样本分开如图,从中可以看出模型的表型也非常好,尤其是在没化疗的样本中,区分的很明显。
进一步的,我们将 是否化疗的病人分开来进行分类,看看模型能不能把样本分开如图,从中可以看出模型的表型也非常好,尤其是在没化疗的样本中,区分的很明显。
 9、ROC分析
9、ROC分析
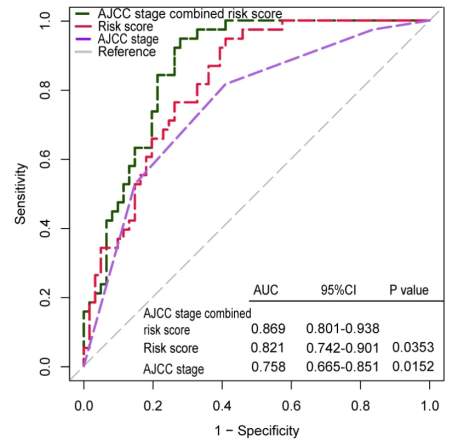
ROC的线下面积能够用来衡量模型的好坏,越大说明模型越好,作者比较了模型的ROC和临床分期Stage的ROC,发现两者的ROC线下面积差不多,如图,但是合并Stage信息和模型得分之后来比较发现具有显著的差异。
未完待续
- 发表于 2018-10-09 09:06
- 阅读 ( 5765 )
- 分类:文献解读
