第三篇lncRNA signature的文章解析,IF五分胶质瘤
这是一篇比较老的建模的文章,发表在Neurobiology of Disease上,目前影响因子五分多点。
Long non-coding RNA expression profiles predict clinical phenotypes in glioma

本文从lncRNA角度出发,分析了lncRNA在胶质瘤中的预后价值,进一步构建了预后模型来对胶质瘤患者进行风险分类,基本流程图如下:
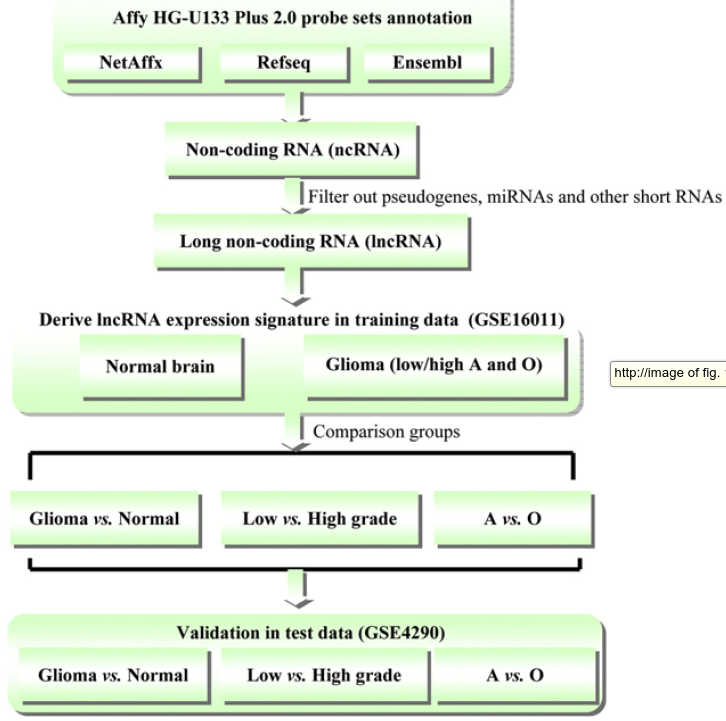
从流程图中可以看出作者首先通过芯片表达谱数据提取lncRNA,然后进行模型构建,最后使用验证数据集进行验证。
下面我们来一步一步的解析一下
1、数据获取
从GEO数据库中下载了GSE16011和GSE4290的原始数据CEL,采用较大的数据集(GSE16011)作为训练集,较小的数据集(GSE4290)作为验证集;
GSE16011共包含268个样本,其中包含13个low-grade astrocytomas (WHO grade 2),175个high-grade astrocytomas (WHO grade 3 and 4),11个low-grade
oligodendrogliomas (WHO grade 2), 69个high-grade oligodendrogliomas(WHO grade 3), as well as 8个non-tumoral brain tissue controls;
GSE4290共包含157个样本,其中包含7个low-grade astrocytomas(WHO grade 2), 100个high-grade astrocytomas (WHO grade 3 and4), 38个low-grade oligodendrogliomas (WHO grade 2), 12个high-
grade oligodendrogliomas (WHO grade 3), and 23个non-tumoral brain controls。
2、数据处理
首先使用RMA对原始CEL数据进行标准化得到探针表达谱
3、lncRNA提取
首先下载NetAffx的注释文件,
一、将探针对应的RefSeq ID上,然后提取出NR_开头的探针,NR代表非编码的,作为预选的lncRNA
二、同时将探针对应ENSG ID上,然后根据Ensembl数据库的基因注释信息,提取属性为:“lincRNA”, “processed transcripts”, “non-coding” or “misc_RNA”的探针,作为预选的lncRNA
三、合并一和二的结果,进一步去除基因属性为:pseudogenes, rRNAs, microRNAs and other short RNAs including tRNAs, snRNAs and snoRNAs
最终得到2448个lncRNA
4、差异分析
作者使用SAM来进行差异的筛选,分析了正常脑组织和其他各个阶段的肿瘤组织的差异,选择阈值为FDR<0.1和Foldchange大于两倍。
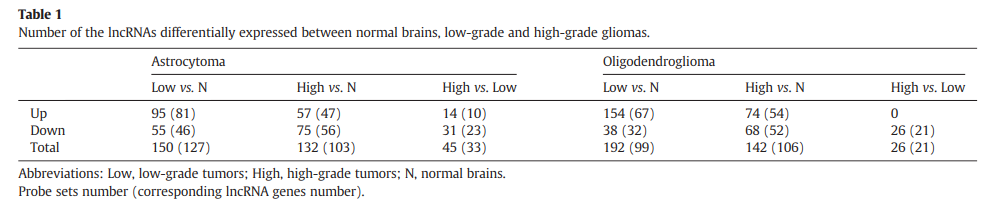
未完待续
- 发表于 2018-10-09 17:06
- 阅读 ( 6217 )
