两篇文章一字之差,影响因子差了三倍,他们都干了啥?(第一篇)
话不多说先上图:

最近在写工具盒里WGCNA工具的使用教程,检索素材时发现了一篇文章,哦不,是两篇,他们长的几乎一样,且看:
Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis.
Prognostic genes of breast cancer revealed by gene co-expression network analysis.
看了大半天愣是没看出差别,最后发现除了首字母大写以外,就差了一个单词:Identified/revealed,这勾起了我的极大兴趣,两篇文章分数都不高,但是相比之下Identified似乎显得牛逼了不少。
看文章先看思路,我们先看一下这个分数低的大概做了什么,从标题Prognostic genes of breast cancer revealed by gene co-expression network analysis可以看出基于权重共表达网络(WGCNA)来揭示了乳腺癌的预后基因,通俗来讲就是使用了WGCNA的方法分析了乳腺癌数据,找到了一些跟乳腺癌预后有关的基因。
先脑补WGCNA方法:通俗点就是:一种根据基因表达水平分析两两基因之间相关性,来对基因进行分类形成不同的模块(共表达模块),这些模块中的基因在不同肿瘤样本的表达变化上非常相似,所以可以推测这些模块中的基因可能共同参与某些功能或与某种表型相关。
他的具体流程是什么样的呢?
首先他在ArrayExpress(和GEO类似的数据库)上找了两套带有预后信息的乳腺癌数据,这两套数据的ID分别为:GSE2034、GSE25066,看了一下这两套数据里的样本分别有286个、508个样本。
然后 下载了这两套数据的芯片原始数据,分别使用RMA进行重新的标准化了一下,然后画了一下标准化后各个样本的表达的箱线图,看看各个样本的数据分布是不是一致如图:

此图长相骨骼清奇,才华内敛,隐约中透露着不凡,从图中我们可以看到A、B图中的样本表达水平大多分布在0-15之间,中位值基本上在一条直线上,所以可以确定我们的样本的表达水平的分布是合适的。
由于我们选择的都是肿瘤样本,没法去做差异分析,也就没法得到差异基因,那么想做功能富集该怎么办呢?思来想去得到妙计一条:我们研究的是预后,也就是说一个基因如果在各个肿瘤样本中表达都没变化那么他跟预后肯定没啥关系,而那些在不同的肿瘤病人样本中表达变化较大的更有可能与病人的预后相关,基于此我们可以通过计算每个基因在各个肿瘤样本变化程度来衡量这些基因的相对重要性,所以我们选择“变异系数”这个指标来计算每个基因在各个肿瘤样本中的重要性,最终我们选择变异系数大于0.5筛选出了2669个基因(主观上觉得0.5这个阈值还行,你觉得不行可以换);
我们得到了这2669个变化较大的基因,我们想看一下筛出来的这些基因都是干啥子的,所以我们拿这些基因做了功能富集分析,发现这些基因它们主要与免疫应答,细胞增殖,细胞分化和细胞粘附相关,这些通路不就是人们常说的与肿瘤的发生发展非常相关的通路嘛,嗯,我想我们筛的这些基因里应该是包含了一些与肿瘤密切相关的基因的,所以下一步我可以根据这些基因的表达水平对这些基因进行分类一下,做个WGCNA,如图:
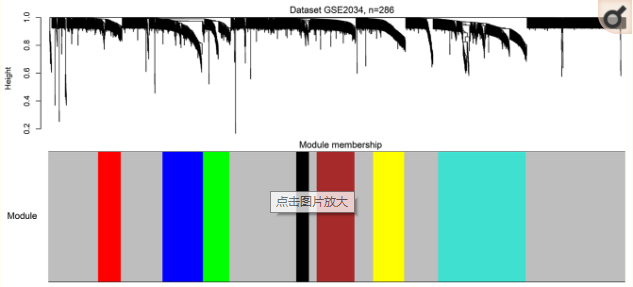
此图依然长相奇特,图中横向的代表基因,纵向的代表基因之间的距离(可以认为是相似度,图中越高表示越不相似),隐约中我们可以看出有八种颜色,他们一簇一簇的存在,这个图告诉了我这些基因可以分为八个模块,其中相同颜色都在一起,这说明这这些同一个模块内的基因他们之间的相关性非常高,那么这里还有一个无处不在的灰色为啥都是穿插着的呢?那是因为灰色是垃圾模块,那些不能正常分类到其他模块中的基因全都丢到灰色的模块里了,所以实际上我们最终得到的是七个有效的模块,所以我们这2669个基因根据他们之间的表达相似程度被分到了这八个模块中,每个模块中的基因他们之间存在紧密联系,我们可以根据这些基因在各个样本中的表达水平来计算出这个模块中的基因整体上的在各个样本中的表达水平我们假定为 特征向量,那么分别计算这八个模块,我们就能得到具有代表模块中所有基因的 八个特征向量,这个时候我们就可以根据这个特征向量结合样本的生存时间来分析一下这几个模块与生存时间的关系了,我们分别计算了两套数据集八个模块的生存时间关系如图:
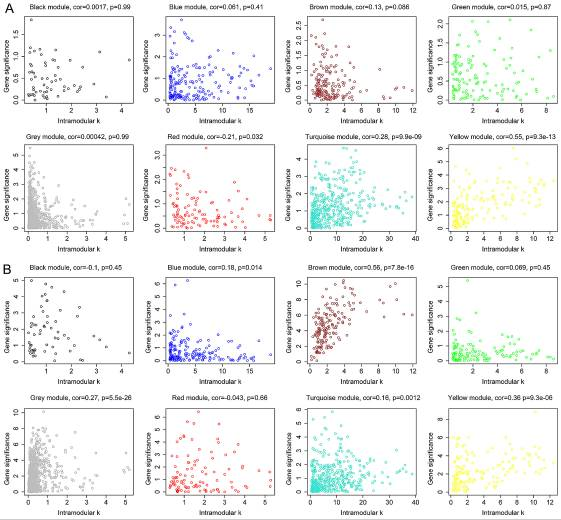 从图中可以看出在两套数据中黄色模块很显眼,他与病人的生存时间呈现明显的正相关性,具有非常高的显著性,9.3×10 -13,9.3×10 -6,这提示了黄色模块中的大部分基因可能与预后密切相关。猜测不是实践,我们进一步的通过单因素生存分析分析了每个模块中的基因与预后的关系,统计显著与预后相关的基因如图:
从图中可以看出在两套数据中黄色模块很显眼,他与病人的生存时间呈现明显的正相关性,具有非常高的显著性,9.3×10 -13,9.3×10 -6,这提示了黄色模块中的大部分基因可能与预后密切相关。猜测不是实践,我们进一步的通过单因素生存分析分析了每个模块中的基因与预后的关系,统计显著与预后相关的基因如图:
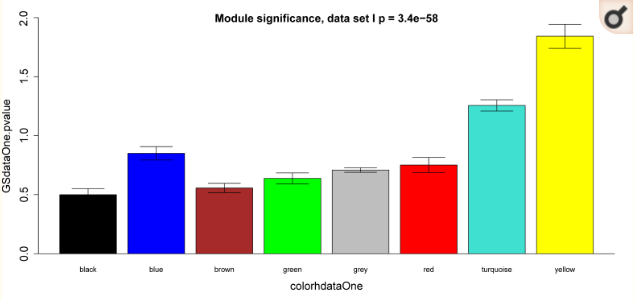 从图中可以看出黄色模块中显著预后相关的基因明显最多,比其他各个模块中的都多。
从图中可以看出黄色模块中显著预后相关的基因明显最多,比其他各个模块中的都多。
到这里我们就可以把焦点挪到黄色模块上了,黄色模块中总共包含了144个基因,我们先看一下这144个基因在各个样本中的表达是什么样本的,根据这144个基因在各个样本中的表达水平线聚个类如图:
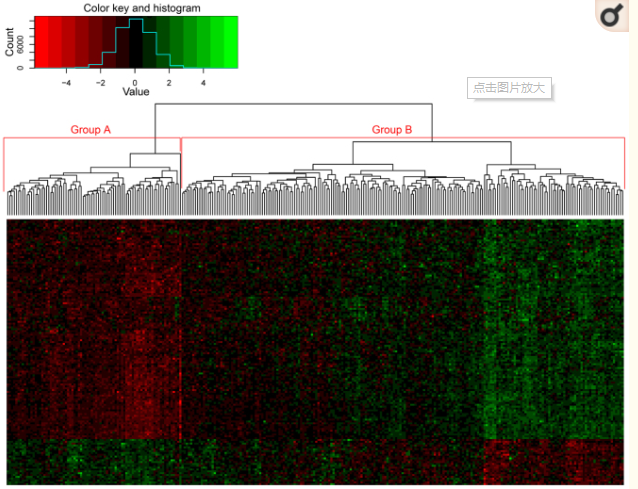 图中横向的代表样本,纵向代表基因,从图中横向的可以看出整体上样本可以分成两组GroupA、GroupB,纵向也差不多能分成两组,一组在GroupA中表达高(图中红色表示低表达),一组在GroupB中表达高,因为我们这个模块中的大多数基因与预后是显著相关的,那么我们有理由猜测GroupA、GroupB两组样本预后有差异,所以我们分析了两组样本的生存时间,发现他们确实存在显著的差异p=0.008,如图:
图中横向的代表样本,纵向代表基因,从图中横向的可以看出整体上样本可以分成两组GroupA、GroupB,纵向也差不多能分成两组,一组在GroupA中表达高(图中红色表示低表达),一组在GroupB中表达高,因为我们这个模块中的大多数基因与预后是显著相关的,那么我们有理由猜测GroupA、GroupB两组样本预后有差异,所以我们分析了两组样本的生存时间,发现他们确实存在显著的差异p=0.008,如图:
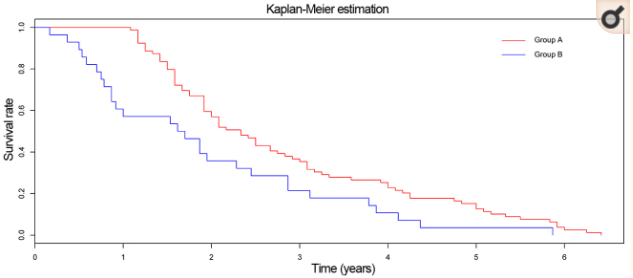 从图中可以看出GroupB的预后显著差于GroupA
从图中可以看出GroupB的预后显著差于GroupA
从表型分析上我们看到了黄色模块的基因与预后密切相关,那么这些基因他们是通过什么途径参与到肿瘤的发展中的呢?我们使用功能富集分析对这黄色模块的144个基因做了KEGG 功能富集分析发现这些基因富集到了细胞周期、卵母细胞减数分裂、p53信号通路、孕酮介导的卵母细胞成熟等生物学通路中,这提示了这些基因可能通过这些通路从而参与到乳腺癌的发展中。
从功能上我们看到了这些基因所富集到的与肿瘤和女性密切相关的通路,那么这144个基因中哪些基因是最具有代表性的呢?我们根据黄色模块的特征向量,分别计算模块中每个基因与模块的特征向量的相关性来筛选与黄色模块最相关的基因(hub gene),最终我们得到了最相关的10个基因:
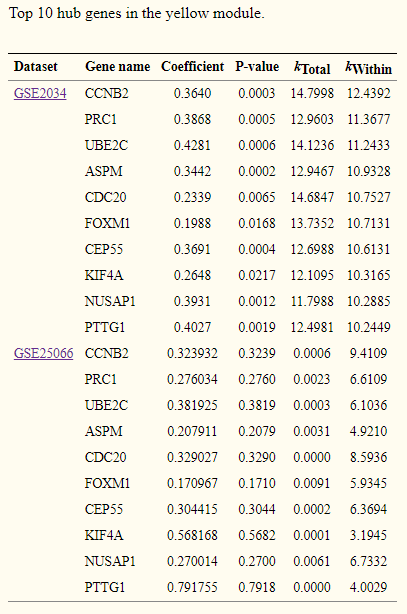 这十个基因其中有几个已经被报道与肿瘤相关,这提示了这十个基因可能是乳腺癌的预后标志物。
这十个基因其中有几个已经被报道与肿瘤相关,这提示了这十个基因可能是乳腺癌的预后标志物。
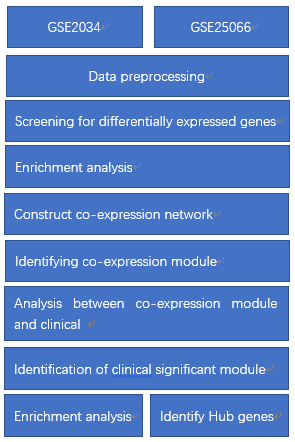
文章到此便结束了,不得不吐槽的是 图太丑了,整个感觉很粗糙,整体流程总结如下:
看第二篇可以移步到这里:两篇文章一字之差,影响因子差了三倍,他们都干了啥?(第二篇)
- 发表于 2018-10-16 16:19
- 阅读 ( 5786 )
- 分类:文献解读
