两篇文章一字之差,影响因子差了三倍,他们都干了啥?(第二篇)
话不多说先上图:

最近在写工具盒里WGCNA工具的使用教程,检索素材时发现了一篇文章,哦不,是两篇,他们长的几乎一样,且看:
Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis.
Prognostic genes of breast cancer revealed by gene co-expression network analysis.
看了大半天愣是没看出差别,最后发现除了首字母大写以外,就差了一个单词:Identified/revealed,这勾起了我的极大兴趣,两篇文章分数都不高,但是相比之下Identified似乎显得牛逼了不少。
看文章先看思路,之前我们看来那篇分数低的,现在我们看一下这个分数稍微高点的大概做了什么,从标题Prognostic genes of breast cancer Identified by gene co-expression network analysis可以看出基于权重共表达网络(WGCNA)来鉴定了乳腺癌的预后基因,通俗来讲就是使用了WGCNA的方法分析了乳腺癌数据,找到了一些跟乳腺癌预后有关的基因。
看第一篇请移步到这里:两篇文章一字之差,影响因子差了三倍,他们都干了啥?(第一篇)
先脑补WGCNA方法:通俗点就是:一种根据基因表达水平分析两两基因之间相关性,来对基因进行分类形成不同的模块(共表达模块),这些模块中的基因在不同肿瘤样本的表达变化上非常相似,所以可以推测这些模块中的基因可能共同参与某些功能或与某种表型相关。
这篇文章提供了流程图如下:
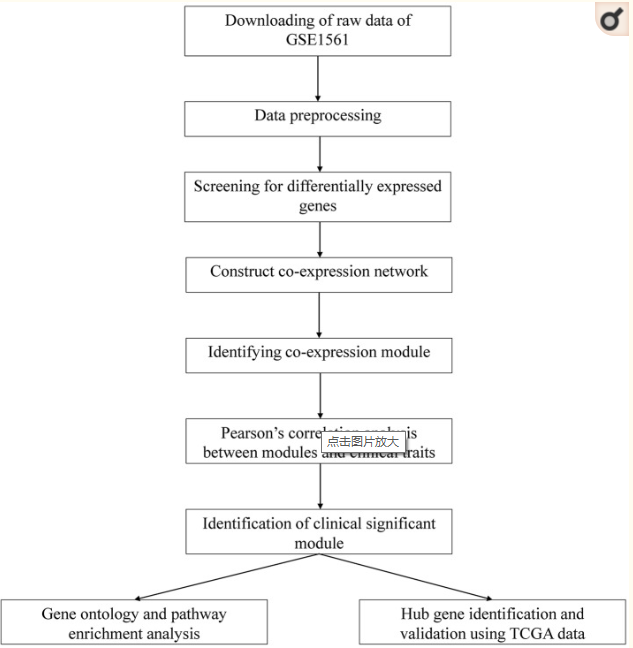 从流程图可以看出:我们先下载了GSE1561的芯片原始数据集,然后重新标准化,然后选择变化大的基因,再进一步做WGCNA分析鉴定共表达模块,再结合临床信息分析模块与临床表型的关系最终筛选出于临床表型密切相关的模块,分析模块中的基因的功能,然后筛选出hub gene,进一步使用TCGA数据集验证。
从流程图可以看出:我们先下载了GSE1561的芯片原始数据集,然后重新标准化,然后选择变化大的基因,再进一步做WGCNA分析鉴定共表达模块,再结合临床信息分析模块与临床表型的关系最终筛选出于临床表型密切相关的模块,分析模块中的基因的功能,然后筛选出hub gene,进一步使用TCGA数据集验证。
那么我们回顾一下前一篇文章的总结性流程图:
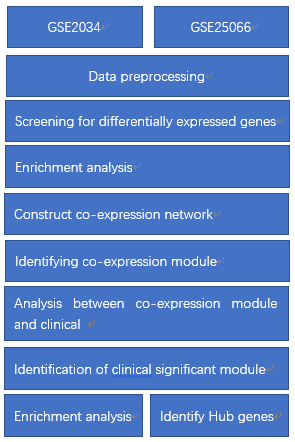
对比发现流程及其相似,在流程上主要差别在于这篇文章用TCGA验证了hub gene,而之前的那篇文章两套数据集并行做的分析,并没有外部数据集作为验证。
我们来一起看一下具体分析上的细节:
首先下载芯片的原始数据,然后使用RMA进行标准化,与第一篇一样
然后筛选变化大的基因,这篇文章中使用“方差”这个指标来分析(这个与我之前写的乳腺癌WGCNA的教程类似),选择方差最大的前50%(主观设定,你随意),最终得到了6,206个基因,第一篇文章使用的是变异系数;
使用这6206个基因做WGCNA分析(这里直接进入主题,而第一篇文章中先做了一下功能富集分析),首先观察各个样本中在这6202个基因的表达水平有没有异常的,所以先做了一下聚类分析,如图:
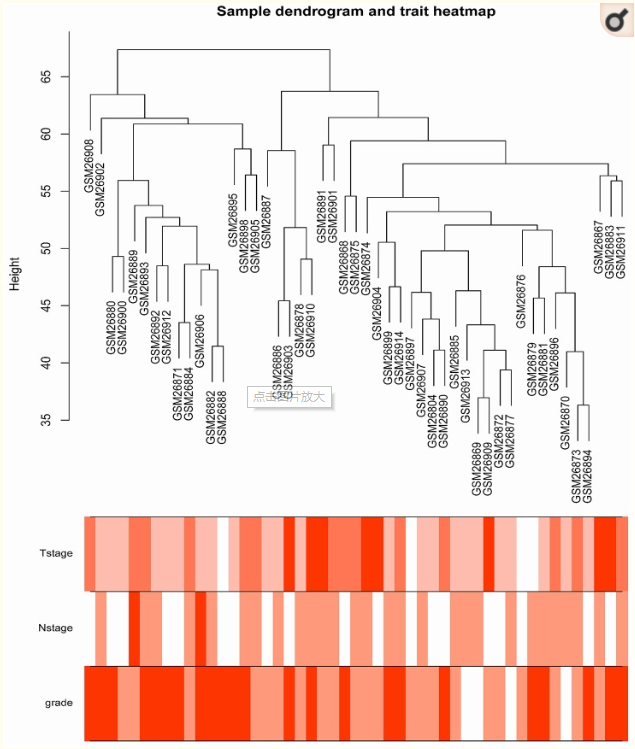 从图中可以看出这些样本之间的并不存在离群的样本,在N分期、T分期和Grade中也没有明显的聚集,所以我们可以放心的拿这些样本进行下一步的分析。
从图中可以看出这些样本之间的并不存在离群的样本,在N分期、T分期和Grade中也没有明显的聚集,所以我们可以放心的拿这些样本进行下一步的分析。
做WGCNA之前需要选择合适的软阈值,我们对1到20的soft-thresholding power进行了网络拓扑分析,并确定了相对平衡的规模独立性和WGCNA的平均连通性。如下图所示,选择阈值9,其为0.9的无标度拓扑拟合指数的最低功率
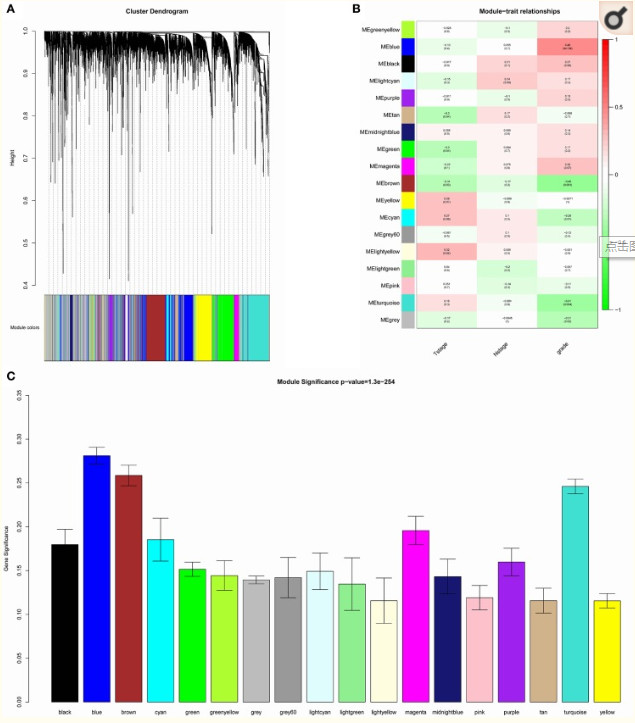
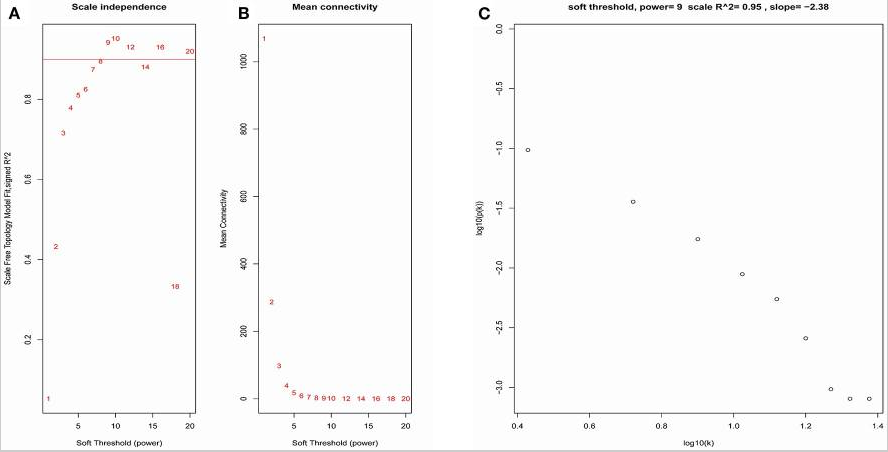 根据软阈值设定为0.9,我们跑了一遍WGCNA,筛选共表达模块,最终得到了18个模块,如下图A,进一步的我们分析了每个模块与临床分期T分期、N分期、Grade的关系如下图B(参考第一篇:每个模块与生存时间的关系),从中可以看出蓝色模块与Grade具有最大的相关性,进一步的我们分析了每个模块中的基因与Grade的相关性如下图C,从中可以看出蓝色模块最高,所以我们锁定了蓝色模块(与第一篇文章中锁定黄色模块的思路完全一样)。
根据软阈值设定为0.9,我们跑了一遍WGCNA,筛选共表达模块,最终得到了18个模块,如下图A,进一步的我们分析了每个模块与临床分期T分期、N分期、Grade的关系如下图B(参考第一篇:每个模块与生存时间的关系),从中可以看出蓝色模块与Grade具有最大的相关性,进一步的我们分析了每个模块中的基因与Grade的相关性如下图C,从中可以看出蓝色模块最高,所以我们锁定了蓝色模块(与第一篇文章中锁定黄色模块的思路完全一样)。
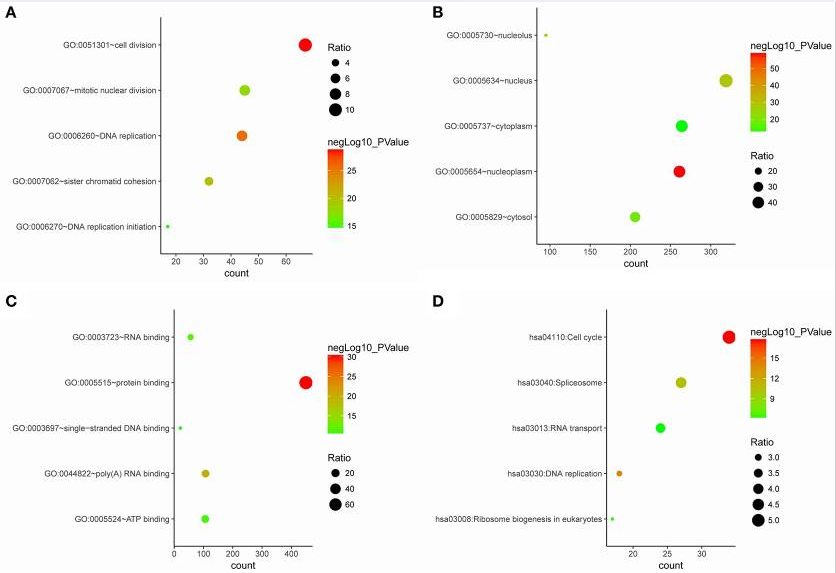
我们将蓝色模块中的基因做GO、KEGG功能富集分析,发现蓝色模块中的基因主要富集到有丝分裂细胞周期过程,这些过程与肿瘤的发生发展密切相关(参考第一篇 黄色模块基因的功能富集分析,思路一致)
 进一步的筛选蓝色模块的hub gene,在第一篇中使用的是top10,而这篇使用的是| MM |> 0.8和| GS |> 0.2(主观设定,你可以适当调整),思路还是类似的,但很明显top10就只剩下10个基因了,而| MM |> 0.8和| GS |> 0.2得到了42个,这给了我们更多的操作空间,我们进一步的分析这42个基因与预后的关系发现 CCNB2, FBXO5, KIF4A, MCM10, and TPX2这五个基因的高表达与差的复发相关,进一步的使用TCGA数据也验证了这一结果。
进一步的筛选蓝色模块的hub gene,在第一篇中使用的是top10,而这篇使用的是| MM |> 0.8和| GS |> 0.2(主观设定,你可以适当调整),思路还是类似的,但很明显top10就只剩下10个基因了,而| MM |> 0.8和| GS |> 0.2得到了42个,这给了我们更多的操作空间,我们进一步的分析这42个基因与预后的关系发现 CCNB2, FBXO5, KIF4A, MCM10, and TPX2这五个基因的高表达与差的复发相关,进一步的使用TCGA数据也验证了这一结果。
到此 两篇文章基本上都是一样的,不同的是第一篇文章到这里故事就讲完了,而这一篇文章从临床亚型上,免疫组化、预后模型上详细的分析了这些hub gene,这便让这篇文章一下子上了一个档次。
首先我们找到了CCNB2, FBXO5, KIF4A, MCM10, and TPX2这五个高表达与差的复发相关的基因,进一步的我们分析了一下这五个基因在不同的亚型中的表达情况如图:
 从图中可以看出这五个基因在不同的Stage中具有显著的差异,在Luminal、Her2、TN分子亚型上也具有显著的差异,尤其是在三阴性乳腺癌中具有显著的高表达,我们都知道三阴性乳腺癌预后最差,这与前面的预后关系相吻合。
从图中可以看出这五个基因在不同的Stage中具有显著的差异,在Luminal、Her2、TN分子亚型上也具有显著的差异,尤其是在三阴性乳腺癌中具有显著的高表达,我们都知道三阴性乳腺癌预后最差,这与前面的预后关系相吻合。
进一步的我们分析了这五个基因在癌与癌旁的表达差异如图:
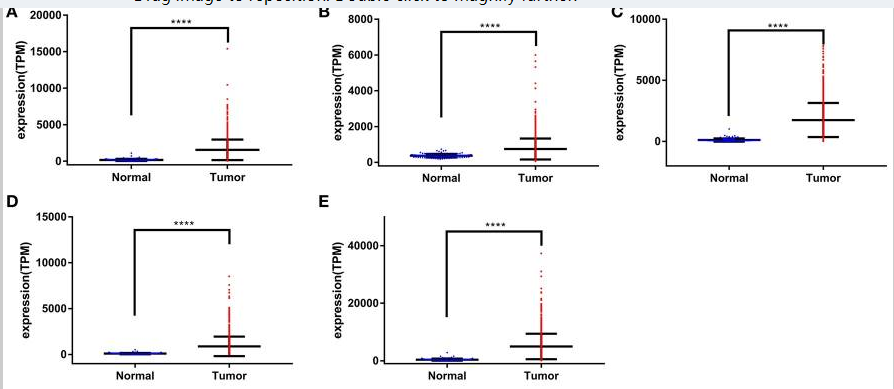
从图中可以看到在肿瘤中都是高表达,这说明这五个基因都是促癌基因,这与他们高表达预后差也吻合;既然他们在癌与癌旁中是差异表达的,那么他们能否作为一个诊断标志物来识别癌症样本呢?我们分别分析了这五个基因作为诊断标志物的可能性,绘制了他们的ROC曲线
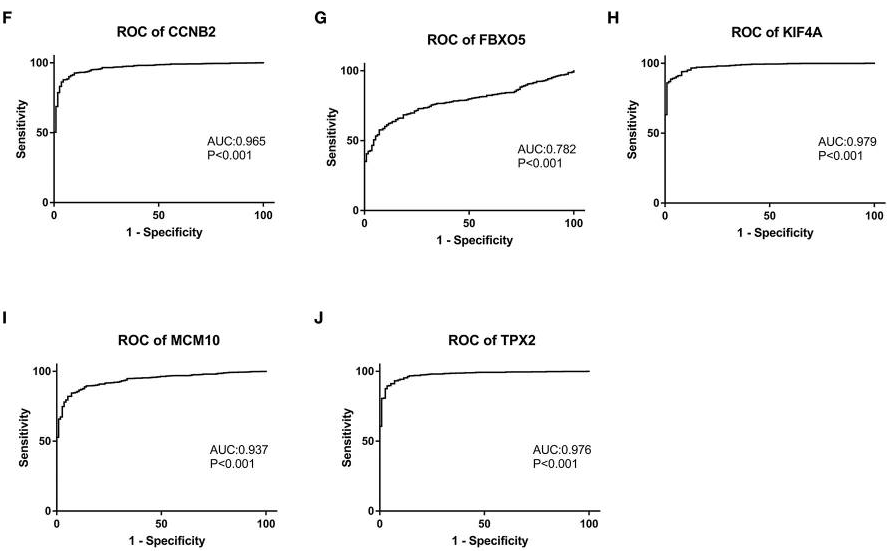
从图中可以看到这五个基因具有很好的诊断作用,AUC线下面积都很高;
到此我们基本上得到了几个结论:
1、这五个基因在肿瘤样本中高表达
2、这五个基因高表达导致肿瘤的预后差
3、这五个基因具有很好的诊断作用
4、这五个基因在不同的分子亚型和病理分析中表达具有显著的差异
我们进一步的从免疫组化实验中再看一下这五个基因在蛋白层面上的表达差异情况,如图:

从中可以看出这五个基因蛋白水平在肿瘤组织中显著高表达。
以上便是文章的全部思路,相比于第一篇,虽然思路相似但是明显精细了很多,更全面。
- 发表于 2018-10-16 16:27
- 阅读 ( 8855 )
- 分类:文献解读
