文献分享(1)肿瘤细胞和正常细胞之间mRNA丰度的关联


肿瘤与环境,两者既是相互依存,相互促进,又是相互拮抗,相互斗争的。它是现代肿瘤生物学的一个关键和核心的问题。近年来由于肿瘤细胞学和分子生物学的进展,人们对于肿瘤和环境的相互关系有了更加深入的了解。这不仅对于认识肿瘤的发生、发展、转移等有着重要的意义,而且对于肿瘤的诊断、防治和预后亦有着重要的作用。
实体瘤是由肿瘤细胞(TC)和正常细胞(TAC)组成的,肿瘤细胞和癌旁细胞的内在的互作形成了肿瘤微环境。但是当前的临床基因组研究是基于组织样本来做的,没有区分是肿瘤细胞的表达还是正常细胞的表达。因此,Natalie S. Fox等人对1780例原发性的乳腺肿瘤的转录组数据进行拆分,也就是将一个组织样本的表达值拆分成两部分表达值,即肿瘤细胞表达值和正常细胞表达值,从而去更好的了解肿瘤细胞和正常细胞之间的互作。在这篇研究中,他们还发现肿瘤细胞和正常细胞的表达和样本的临床表型(疾病亚型和病人的生存)很相关。并且还揭示了肿瘤细胞和正常细胞之间独特的驱动基因突变模式(比如TP53)。通过Natalie S. Fox等人的方法,进一步阐明了乳腺肿瘤内部的分子相互作用和这种微环境条件下去带来的肿瘤表型(如图Fig 1)。
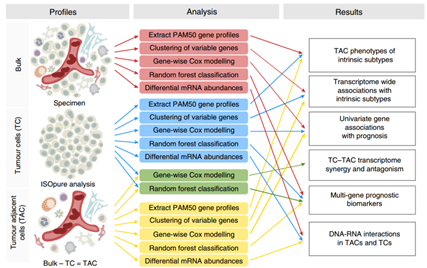 Fig 1. 分析流程图
Fig 1. 分析流程图
今天小编为大家带来这篇文章正是来探讨这一问题的
标题:Landscape of transcriptomic interactions between breast cancer and its microenvironment
杂志:Nature Communication IF:11.878 发表时间:2019.7.15

(1)数据:METABRIC数据集mRNA以及突变SNV数据(https://www.ebi.ac.uk/ega/datasets/EGAD00010000162 & https://github.com/cclab-brca);TCGA乳腺癌数据(https://gdac.broadinstitute.org/)。
(2)对mRNA表达谱数据进行log2转化,并通过ISOpureR[1]算法计算样本的肿瘤纯度,公式如下:
b代表组织样本的mRNA表达值,t代表ISOpureR算法得到的肿瘤细胞mRNA表达值,p代表ISOpureR算法估计样本是肿瘤的概率值,s代表正常细胞mRNA表达值。其中,s就是基于该公式想要去求的变量。
(3)识别差异mRNA:R语言limma包。
(4)亚型聚类:基于那些在组织样本、肿瘤细胞mRNA表达或是正常细胞mRNA中表达标准差大于1的基因,通过R语言ConsensusClusterPlus包对样本进行聚类。
(5)通路富集分析:通过网页工具http://biit.cs.ut.ee/gprofiler/[2]对基因进行功能富集分析。
(6)基因对分析:高表达mRNA指在肿瘤细胞表达谱中高表达且在正常细胞表达谱中低表达的那一类mRNA;低表达mRNA指在肿瘤细胞表达谱中高表达且在正常细胞表达谱中低表达的那一类mRNA或者在肿瘤细胞表达谱和正常细胞表达谱中都低表达的mRNA。
(7)构建预后分类模型:通过RandomForest算法来实现预后分类模型的构建。先将样本分成训练集和测试集,对于训练集而言,循环5000次,每次挑选50个基因构建预后分类模型,并通过在测试集通过计算曲线下面积(AUC)对模型进行验证。

(1)分解得到肿瘤细胞和正常细胞表达谱:基于乳腺癌组织表达谱数据,通过ISOpure方法,结合方法中的公式,进而得到肿瘤细胞和正常细胞的mRNA表达谱。文章中,作者还考虑到乳腺癌是一个高度异质性的疾病,还通过PAM50识别样本亚型,发现将样本分亚型去计算mRNA表达和不分亚型去计算mRNA表达的相关性高(如Fig 2b,2c所示)。同时,为进一步验证模型的有效性,还在各亚型分解表达谱中去看ESR1,PGR以及ERBB2三个乳腺癌重要的免疫组化检测蛋白的表达情况(如Fig 2d)。CAV1[3,4]是乳腺癌的预后相关基因,在分解得到的正常细胞表达谱中,将CAV1表达的中位数值作为阈值,将样本分为高地表达组,看两组生存情况的差异(如Fig 2e)。
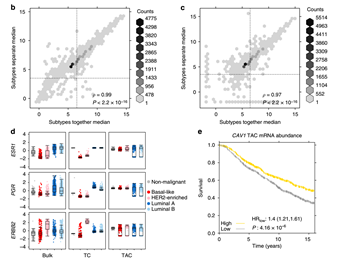 Fig 2. 分解算法有效性评估
Fig 2. 分解算法有效性评估
(2)亚型表达分析:使用PAM50算法,对肿瘤组织样本表达谱、肿瘤细胞表达谱以及正常细胞表达谱中的样本识别亚型,并得到对应亚型下这些病人50个基因的表达均值(如Fig 3a)。接下来,基于高变异(在任一表达谱中标准差的值大于1)的基因进行聚类,比较和PAM50亚型划分的样本类的一致性,发现正常细胞mRNA表达的峰度明显不同于肿瘤细胞表达谱中的(如Fig 3b-3e)。这些结果说明了正常细胞表达谱和乳腺癌亚型的临床表型相关
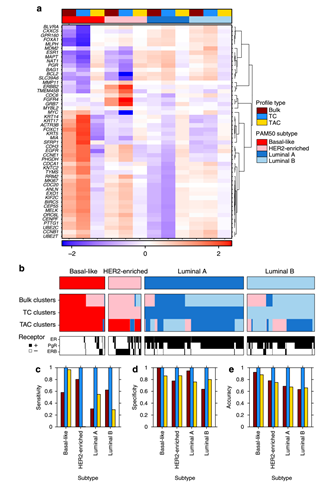 Fig 3. 不同亚型mRNA表达峰度可视化
Fig 3. 不同亚型mRNA表达峰度可视化
(3)预后分析:想进一步去验证在分解得到的肿瘤细胞表达谱和正常细胞表达谱中预后基因是否差异,因此,采用。和组织样本比较,分解得到的肿瘤细胞表达谱与之共享有和好的预后和坏的预后相关的基因相关的基因(如Fig 4b)分解得到的正常细胞表达谱与之共享有与好的预后和坏的预后相关的基因相关的基因(如Fig 4c)。分解得到的正常细胞表达谱和肿瘤表达谱之间有共享的和预后相关的基因(如Fig 4d)。分解得到的肿瘤细胞表达谱和正常细胞表达谱得到的预后相关的基因与组织样本表达谱得到的预后基因具有一致性,并且这些基因是亚型特异的。
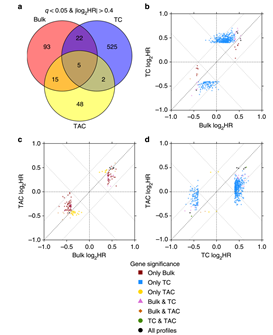 Fig 4. 预后相关基因的展示
Fig 4. 预后相关基因的展示
(4)分解的肿瘤细胞与正常细胞表达谱间基因的协同与拮抗作用:对于每个基因来说,计算其分别在组织、分解的肿瘤细胞和正常细胞表达谱之间的表达中位数值,根据其值的大小进行组合,对基因进行不同的分组,并进一步分析其对预后的影响,如Fig 5所示,发现组织表达谱可能会掩盖一些重要信息,反而是将组织样本表达谱进行分解后更能找到和病人预后相关的基因。
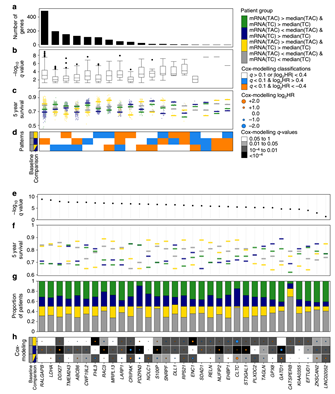 Fig 5. 分解的肿瘤细胞与正常细胞表达谱基因互作关系
Fig 5. 分解的肿瘤细胞与正常细胞表达谱基因互作关系
(5)预后性能的评价:将1780个样本分成训练集(n = 1430)和测试集(n = 350),通过随机森林,每次随机挑选50个基因来去构建模型,循环5000次,计算每一次的曲线下面积(AUC),发现对预后的预测能力,从大到小排的话,顺序是:分解的肿瘤细胞表达谱、组织数据表达谱以及分解的正常细胞表达谱。在Fig 6a和6b中展示了基于不同表达谱构建的分类器的预测准确率,Fig 6c和6d展示了相应的生物标志物对预后的影响。
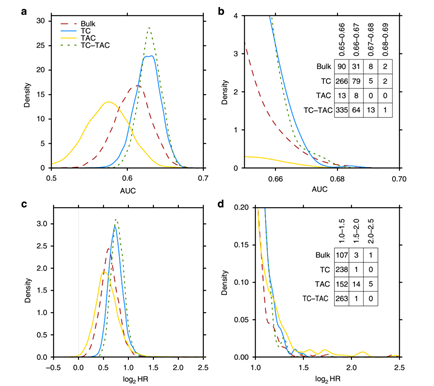 Fig 6. 随机森林模型对分解表达谱预后性能的评估
Fig 6. 随机森林模型对分解表达谱预后性能的评估
(6)突变对mRNA表达峰度的影响:结合在乳腺癌中突变的基因数据[5],基于limma包,首先找到在分解的肿瘤细胞与正常细胞表达谱中差异表达的mRNA,接下来,结合乳腺癌的SNV数据去看这些差异mRNA是否发生非沉默突变。进一步地,考虑到乳腺癌PAM50亚型,在不同的亚型中去刻画其基因突变的情况(如Fig 7a)。在HER2-enriched亚型病人中,对与TP53突变相关的差异mRNA进行分析和可视化(如Fig 7b),也去分析了在luminal亚型中和CDH1突变相关的差异mRNA进行分析和可视化(如Fig 7c)。
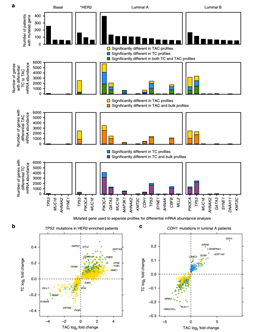 Fig 7. 突变对mRNA表达峰度的影响
Fig 7. 突变对mRNA表达峰度的影响

肿瘤是由良性和恶性细胞组成的复杂混合物,这两类细胞共同构成了相互关联的肿瘤微环境。在肿瘤研究中,只有分析多个细胞群的组合才能去真正了解肿瘤临床表型的进展和起源。在这篇文章中,作者去刻画了组织样本、分解后的肿瘤细胞和正常细胞之间mRNA丰度的关联,发现使用分解后的肿瘤细胞mRNA表达谱较组织样本的mRNA表达谱去对肿瘤进行研究,前者能识别更鲁棒的基因标志物。总结来说,就是通过计算机模拟分解的肿瘤细胞和正常细胞mRNA表达有利于肿瘤之间复杂的转录网络、微环境和癌症侵袭的研究。

1. Quon, G. et al. Computational purification of individual tumor gene expression profiles leads to significant improvements in prognostic prediction. Genome Med. 5, 29 (2013).
2. Reimand, J. et al. g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W89–W89 (2016).
3. Sloan, E. K. et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am. J. Pathol. 174, 2035–2043 (2009).
4. Witkiewicz, A. K. et al. Towards a new ‘stromal-based’ classification system for human breast cancer prognosis and therapy. Cell Cycle 8, 1654–1658 (2009).
5. Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016).

文献分享(3)sEV相关基因表达特征(3-PPI-Mod)与肿瘤内缺氧状态显著相关???
文献分享(4)联合抑制PD-L2和TGF-β2可能会提高免疫治疗的效果?
文献分享(6)对表观遗传酶进行综合泛癌分析揭示了癌症中表观基因组失调的普遍模式
文献分享(7)circRNA特征预测II / III期结肠癌术后复发
- 发表于 2019-11-25 18:46
- 阅读 ( 6549 )
- 分类:文献解读
