五彩进化树与热图更配-ggtree美颜进化树(宏基因组扩增子)
研究基因功能的人建个树,需要找近缘物种、外类群十几至几十个物种,费N天的劲才能做个树。而宏基因组领域的人不用去收集其它物种,因为研究的对像本身就有几百到几千的物种,为了方便阅读或展示主要信息,我们反而会挑选结果中前100以内的物种去分析并展示。这是我们绝对的优势。
- 4
- 5
- 刘永鑫
- 发布于 2017-10-13 20:05
- 阅读 ( 13967 )
生信人值得拥有的编程模板-Perl
为什么要学编程 图1. 重复工作任务量与时间关系[1] 如上图,对于大量重复工作,非编程者(non-geek)工作量和时间是正相关的,就像富士康流水线上的工人,这种工作对于高智商的人是无法忍受(富...
- 3
- 4
- 刘永鑫
- 发布于 2017-10-13 20:03
- 阅读 ( 5146 )
PICRUSt:16S预测宏基因组-扩增子分析锦上添花
16S分析能获得的信息比较有限,一般找到差异OTU,就很难再深入分析了。 如何把差异OTU与细菌自身的基因组功能建立联系呢?很多人在这方面做出了努力。
- 2
- 7
- 刘永鑫
- 发布于 2017-10-13 20:02
- 阅读 ( 8958 )

你还不会安装Circos?其实就三步!!!
本文内容首发于【计算表观遗传学】公众号,对表观遗传学领域感兴趣的朋友可以移步了解更多。如有疑问可留言交流。
- 1
- 5
- Jason Y.J. Wei
- 发布于 2017-10-13 16:55
- 阅读 ( 8586 )
植物MWAS研究—谷子产量与微生物组关联分析
本文内容首发植物微生物组公众号,更多阅读欢迎跳转植物微生物组公众号 MWAS简介 微生物组关联分析(Microbiome/Metagenome-wide association studies , MWAS)是指捕获多维尺度上的互作作用,...
- 2
- 4
- 刘永鑫
- 发布于 2017-10-13 11:59
- 阅读 ( 6800 )
Nature:拟南芥微生物组功能研究1培养组学—高通量细菌分离培养鉴定
本网对Markdown排版支持较差,请跳转植物微生物组公众号阅读 背景介绍 在近年来,随着宏基因组技术的发展,对原核生物的纯培养逐渐被忽视,但纯培养对于阐明这些微生物的功能是必需的。 本...
- 2
- 5
- 刘永鑫
- 发布于 2017-10-13 11:57
- 阅读 ( 12159 )
Endnote X8云同步:家里单位实时同步文献笔记,有网随时读文献
本文适合已经在使用Endnote X8的用户。什么?你连软件都没用过,赶快后台回复“endnote”下载最新版。基本操作也不会用吗?
- 2
- 4
- 刘永鑫
- 发布于 2017-10-13 11:55
- 阅读 ( 5242 )
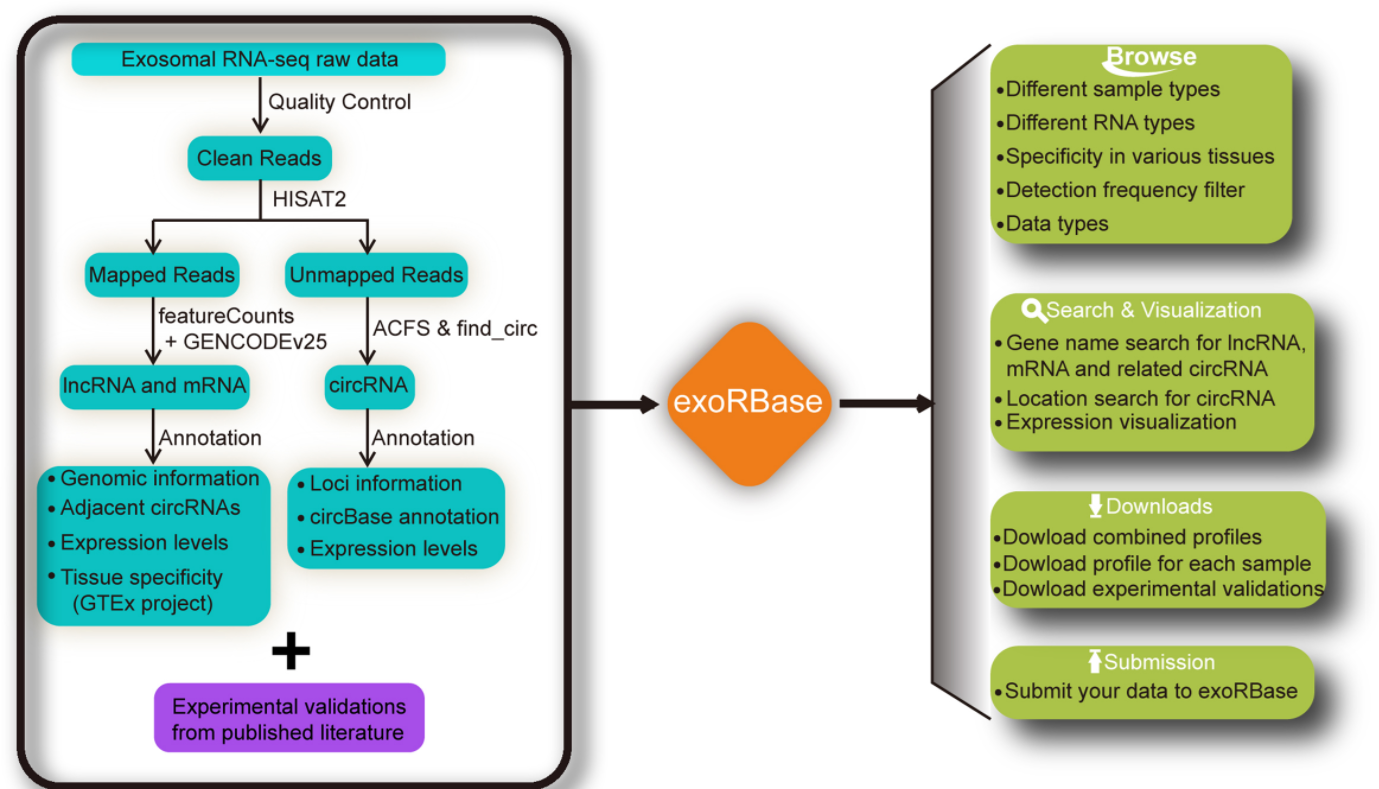
exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes
exoRBase一个整合血液外泌体RNA的数据库。
- 1
- 4
- 张海伦
- 发布于 2017-10-13 09:51
- 阅读 ( 7075 )
你想要的宏基因组-微生物组知识全在这
本文转载自“宏基因组”,更多文章跳转公众号阅读 宏基因组/微生物组是当今世界科研最热门的研究领域之一,为加强本领域的技术交流与传播,推动中国微生物组计划发展,中科院青年科研人员创立“...
- 2
- 5
- 刘永鑫
- 发布于 2017-10-12 23:03
- 阅读 ( 4213 )
扩增子分析神器USEARCH简介
本文中引用统计采用Google学术,统计日期截止2017年10月9日。 Usearch简介 主页:http://www.drive5.com/usearch/ Usearch是什么?它是超快的序列分析软件,在序列比对、聚类、操作等多领域...
- 3
- 4
- 刘永鑫
- 发布于 2017-10-12 23:01
- 阅读 ( 7804 )
扩增子分析还聚OTU就真OUT了,试试unoise3
宏基因组领域是当今热门领域,也正是方法快速发展和变革的时代。之前还把 97%聚类OTU作为扩增子行业的金标准。转眼间各位大佬纷纷向OTU聚类方法拍砖,都不建议再使用。 Feature代替OTU是趋势...
- 2
- 4
- 刘永鑫
- 发布于 2017-10-12 23:00
- 阅读 ( 7530 )

宏基因组分析教程-Analysis of Metagenomic Data
加拿大安大略研究所建立的生物信息网(https://bioinformatics.ca/),每年组织多场生物信息学线下培训,并线上共享培训资料。有超多生信学习PPT和视频,每一份都价值不菲。2016年6月22-24日,在温哥华举办了宏基因组数据分析专题培训,我们可以点击主页中宏基因组专题栏目的 "Access Content" 访问这次培训的材料。
- 3
- 6
- 刘永鑫
- 发布于 2017-10-12 21:59
- 阅读 ( 9661 )
社区打赏功能使用教程
众人拾柴火焰高,为了鼓励广大生信爱好者分享知识,社区最新系统中增加了打赏功能以资鼓励生信人分享有用的知识。 打赏功能设置如下: 首先在账号设置(https://www.shengxin.ren/profile/bas...
- 0
- 3
- admin
- 发布于 2017-10-12 09:27
- 阅读 ( 11162 )
在R语言中关于目录的问题小结
在敲了很多R脚本,然后需要互相引用的时候就会遇到路径问题了 1、如果获取当前工作目录使用: getwd() 2、获取R 包安装目录使用: .libPaths()[1] 3、获取R脚本当前目录咋整? 这个问题其...
- 1
- 0
- zlf
- 发布于 2017-09-30 15:13
- 阅读 ( 4827 )
火山图绘制工具使用说明
本工具专门用来绘制火山图,可以自由定制颜色,横轴纵轴的最大最小值,导出发表级的图片。 软件输入只需两列,一列为foldchang(有没有做log2转换即可),一列为p值(有没有log皆可) 软件界...
- 2
- 7
- zlf
- 发布于 2017-09-29 10:28
- 阅读 ( 14896 )
如何极其简单的使用GEO数据来做差异分析
无论你是要看某个基因是否差异表达或者筛选某个GEO数据集的差异基因,这个方法绝对能够帮助你事半功倍 首先假设你已经找到了一套数据GSE32323 这套数据共包含44个样本,其中有17个配对的癌与...
- 52
- 31
- zlf
- 发布于 2017-09-28 10:48
- 阅读 ( 77351 )
这应该是史上最简单好用的GSEA富集分析工具了
GSEA是什么,这个请自行百度,在使用之前要知道GSEA是啥 然后上工具截图: 一般情况下咱们要做GSEA需要准备三个文件 1、gct格式的表达谱 2、分组文件 3、富集的背景文件,也就是gmt格式的...
- 18
- 25
- zlf
- 发布于 2017-09-19 10:12
- 阅读 ( 65378 )
记一次ORFfinder本地化所遇到的问题
本地化ORFfinder本来是一件相当简单的事情,NCBI已经做的足够简洁,下载ORFfinder二进制文件,然后就可以了,但是我这边却遇到了难题。
- 0
- 1
- deepxin
- 发布于 2017-09-18 19:47
- 阅读 ( 4870 )