TCGA下载工具超级版,在线秒下载
终于TCGA数据下载的终结版上线了,下载地址如:http://sangerbox.com/TcgaDown 界面沿用了之前的风格,一张图秒懂: 每种数据类型下载到个人中心后可以看到 合并的数据 和 单个的原始TCGA下...
- 0
- 4
- zlf
- 发布于 2020-04-16 20:40
- 阅读 ( 7704 )
在GEO数据库如何找带有预后信息的数据?一分钟搞定,适用于找任何的样本特征数据
GEO数据库是目前基因表达谱数据数据库之一、里面包含了各种物种的各种测序和芯片平台的数据。详情介绍可以参考这篇文章:https://shengxin.ren/article/454,在这里我们主要介绍怎么快速的获取 ...
- 12
- 7
- zlf
- 发布于 2020-02-07 10:32
- 阅读 ( 21192 )
怎样使用SangerBox进行投稿选刊
怎样使用SangerBox进行投稿选刊 怎样使用SangerBox进行投稿选刊 怎样使用SangerBox进行投稿选刊 怎样使用SangerBox进行投稿选刊
- 0
- 2
- zlf
- 发布于 2019-12-17 15:16
- 阅读 ( 5173 )
怎样使用Sangerbox对搜索结果进行统计
怎样使用Sangerbox对搜索结果进行统计 怎样使用Sangerbox对搜索结果进行统计 怎样使用Sangerbox对搜索结果进行统计 怎样使用Sangerbox对搜索结果进行统计
- 0
- 0
- zlf
- 发布于 2019-12-17 15:15
- 阅读 ( 3407 )
怎样利用Sangerbox收藏文献,高效读文章
怎样利用Sangerbox收藏文献,高效读文章 怎样利用Sangerbox收藏文献,高效读文章 怎样利用Sangerbox收藏文献,高效读文章 怎样利用Sangerbox收藏文献,高效读文章
- 0
- 0
- zlf
- 发布于 2019-12-17 15:07
- 阅读 ( 3483 )
怎么高效的使用Sangerbox查文献
怎么高效的使用Sangerbox查文献 怎么高效的使用Sangerbox查文献 怎么高效的使用Sangerbox查文献 怎么高效的使用Sangerbox查文献
- 0
- 0
- zlf
- 发布于 2019-12-17 15:05
- 阅读 ( 3834 )
如何快速的查询某学者的基金项目列表
用Sangerbox公众号,便捷快速。 近日公众号上线了新功能,可以根据人名或者人名+单位 就能检索此人的所有基金项目哦 此外还能预测与此人的亲密合作伙伴呢
- 0
- 72
- zlf
- 发布于 2019-12-13 11:07
- 阅读 ( 26063 )
你知道桑格组手公众号怎么查询即时影响因子了吗
好久没有更新公众号的杂志影响因子查询的功能了,最近做了一下改版,主要有两个方面: 1、优化了搜索方式,之前是需要输入完整的杂志名称才能查影响因子及分区,现在支持模糊搜索啦,比如搜索p...
- 0
- 78
- zlf
- 发布于 2019-11-26 20:00
- 阅读 ( 117634 )
如何在 Linux 中查看目录大小?
这是一篇关于如何通过一些常用的命令,显示 CentOS 或 RedHat 中的 Linux 目录大小,以及哪些文件夹占用的空间最大的教程。 搜索当前的 CentOS 或 RedHat 文件夹 您可以使用以下命令,在命令行中??
- 0
- 0
- 史提芬先森
- 发布于 2019-11-26 14:06
- 阅读 ( 3954 )

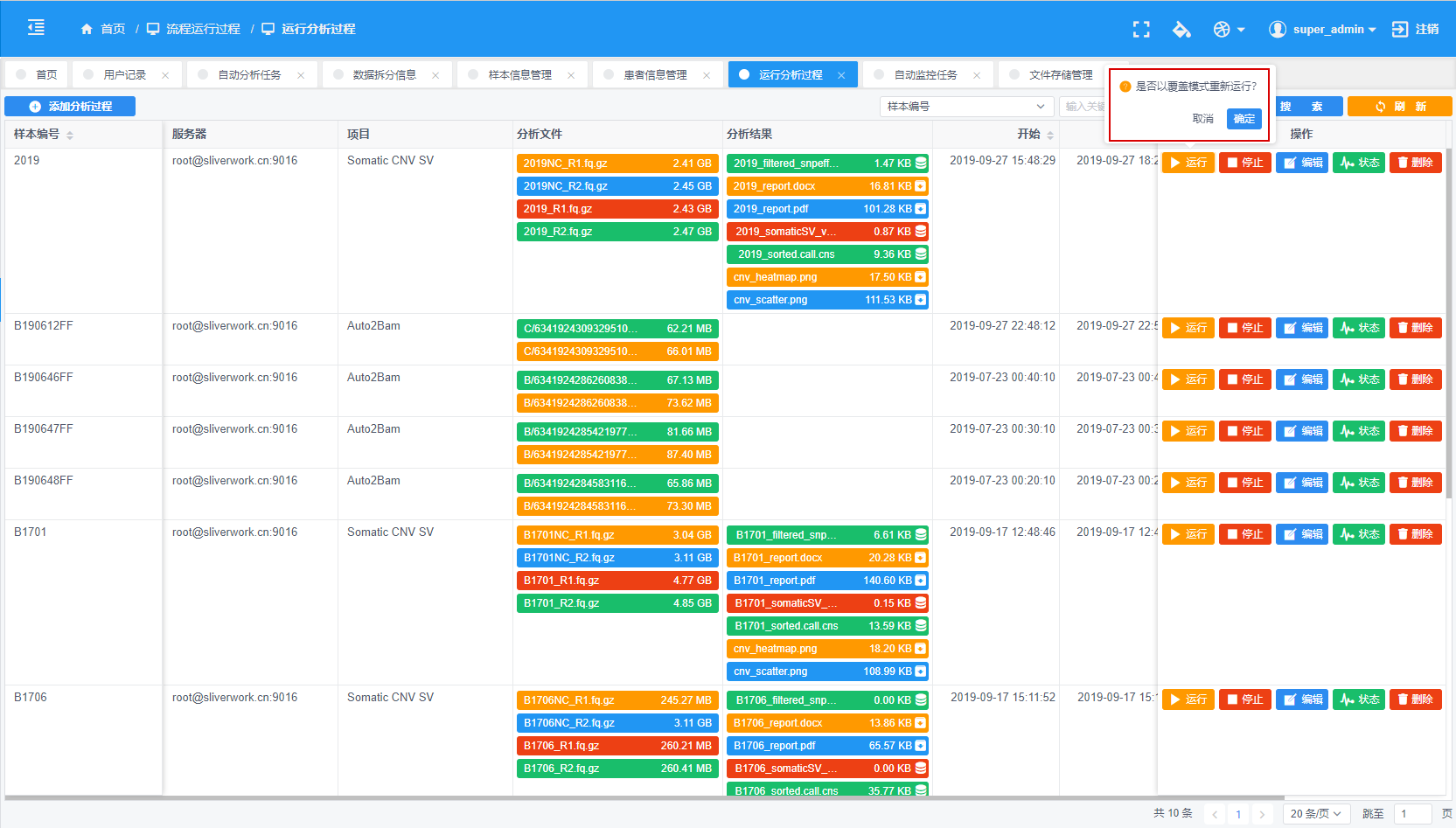
生信分析云平台产品开发 - 6 生信分析pipeline批量运行与过程控制
一、分析流程的批量运行顺序 二、分析流程的过程控制
- 0
- 0
- 豆浆包子
- 发布于 2019-10-14 12:28
- 阅读 ( 3996 )
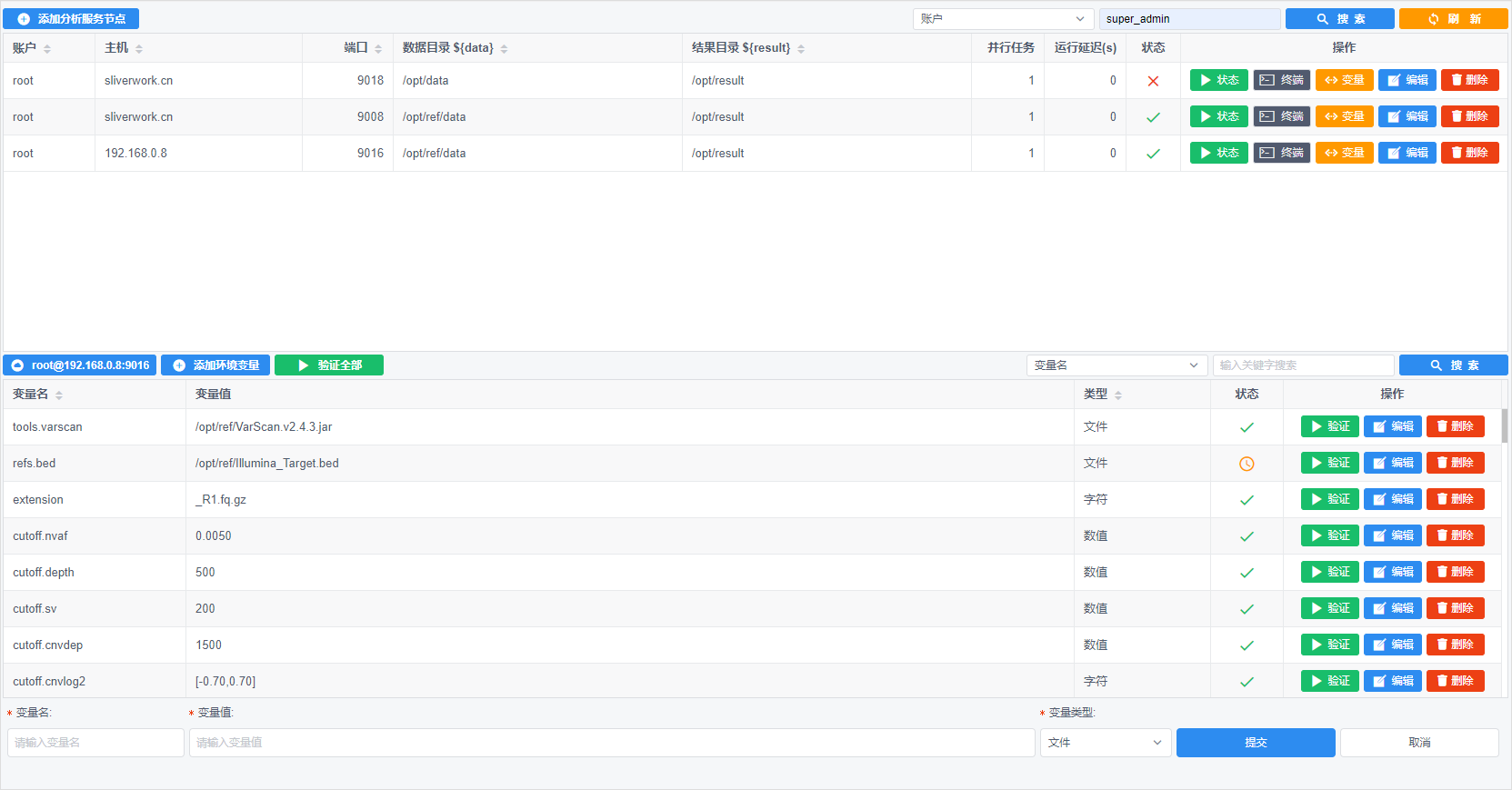
生信分析云平台产品开发 - 5 生信分析pipeline服务器端运行
前文链接: 图形化生物信息分析系统开发 - 1 需求分析及技术实现 图形化生物信息分析系统开发 - 2 样本信息处理 图形化生物信息分析系统开发 - 3 生信分析pipeline的进化 图形化生物信息分...
- 0
- 0
- 豆浆包子
- 发布于 2019-09-27 14:23
- 阅读 ( 3566 )

生信分析云平台产品开发 - 4 生信分析pipeline图形化
变量处理:要实现pipeline图形设计器,首先要先对用到的变量,做统一的设计 分析步骤/节点设计:前文提到,生信分析pipeline其实就是基于文件输入输出的工作流,这里对工作流做了简化,归纳起来工作流中有4种节点。 工作流设计: 最后,有了变量,和节点,最后就是工作流的设计了。用连接线,将以上4中节点连接起来,计算相互之间的依赖关系,用统一的格式保存起来。
- 0
- 0
- 豆浆包子
- 发布于 2019-09-19 13:23
- 阅读 ( 5228 )
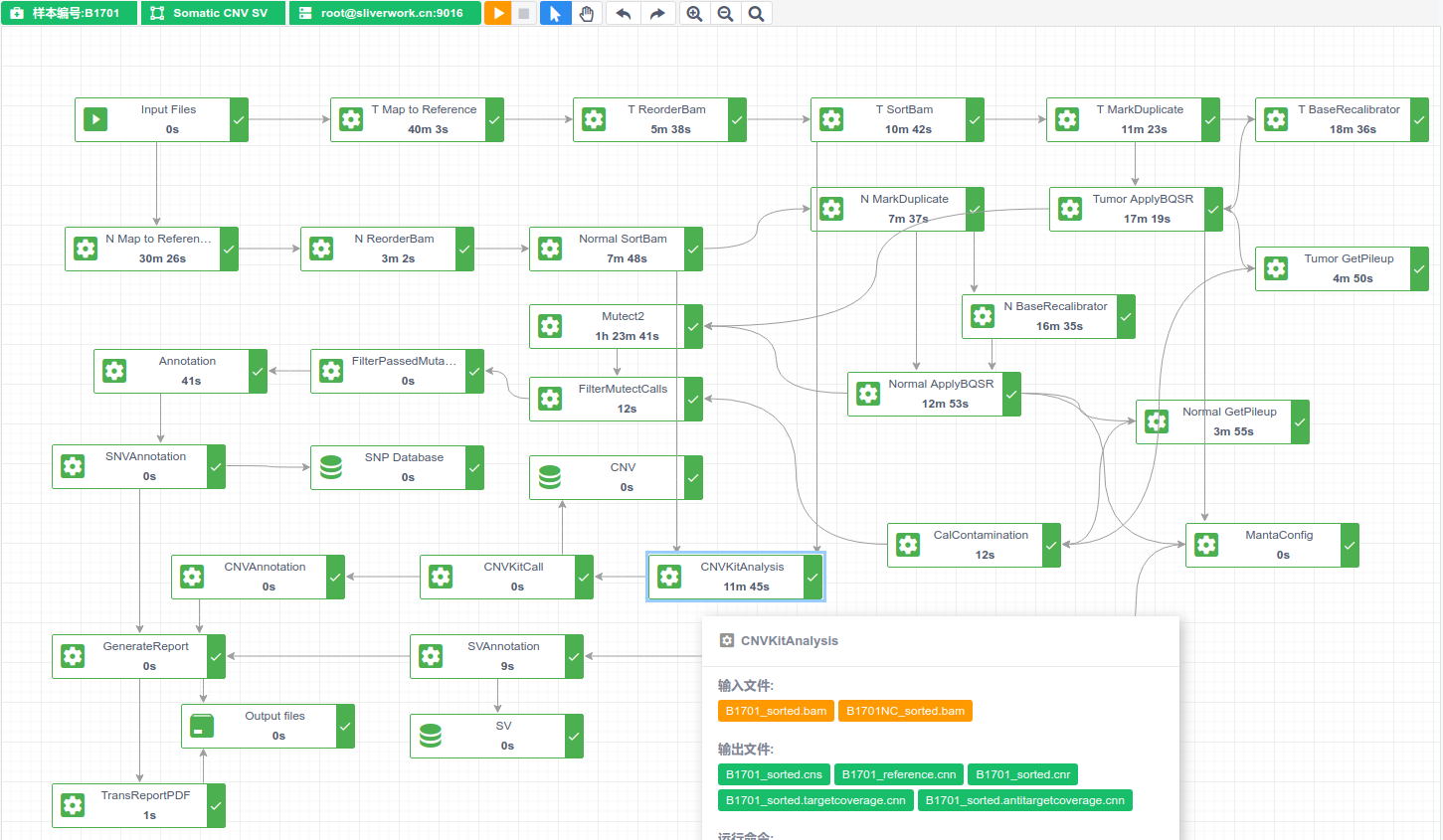
生信分析云平台产品开发 - 3 生信分析pipeline的进化
接上两篇内容,本文主要讲述工作中NGS从科研进入医学临床领域,工作中接触到生信流程,以及最终在自动图形化开放式生信分析系统开发中生信workflow设计实现的过程。 1.手动命令行运行 2. 脚本连续运行 3.一个脚本 shell 文件运行整个分析流程 4. 自动扫描文件并运行脚本 5. 带报告的自动扫描并触发运行脚本
- 0
- 0
- 豆浆包子
- 发布于 2019-09-09 18:19
- 阅读 ( 6448 )

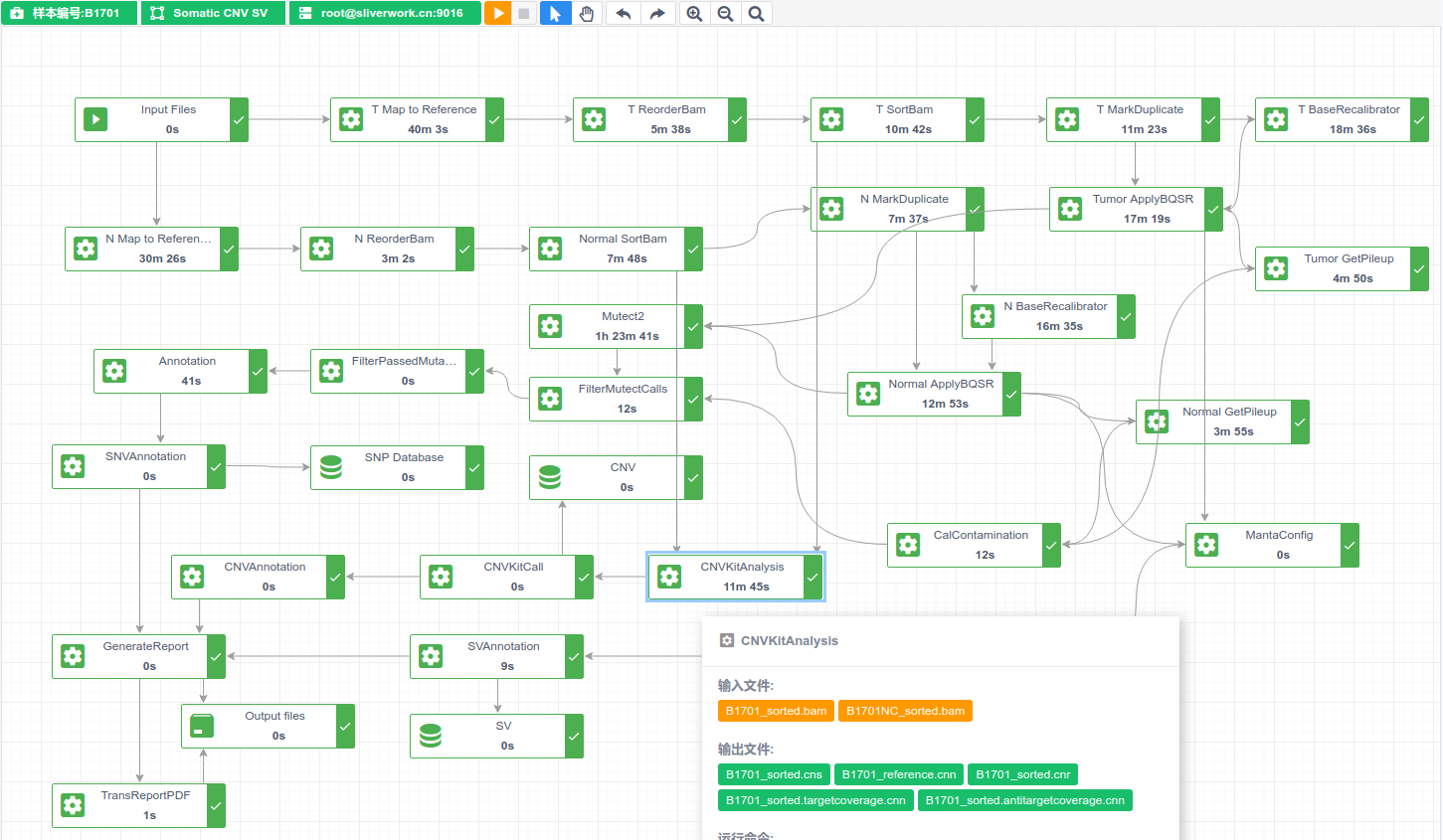
生信分析云平台产品开发 - 1简单需求分析及技术实现
我的生信生产系统/全自动分析系统开发过程- 1简单需求分析及技术实现 起因/背景 几张图片 实践问题一,图形化替代命令行脚本交互 实践问题二,解决迁移部署问题 实践问题三,解决环境搭建、软件安装问题 需求:分析流程(pipeline)能够快速部署迁移 技术实现:使用虚拟化技术: 最终软件架构设计如图: 自动运行结构如图(针对Illumina机型):
- 0
- 2
- 豆浆包子
- 发布于 2019-08-21 17:19
- 阅读 ( 6115 )
使用sigclust来评估你的聚类是否显著
看到文献中(doi:10.1080/2162402X.2017.1392427)有人使用sigclust来评估聚类是否合理,于是乎研究了这个包。 sigclust主要针对生物信息学数据(高维低样本)产生的聚类结果进行评估,一个基本假...
- 1
- 1
- zlf
- 发布于 2019-02-23 14:36
- 阅读 ( 6939 )
TCGA简易下载工具V16版升级详细教程
主要优化了三个方面: 一、miRNA数据合并时同时合并出两个矩阵,一个是counts的,一个是FPKM的 二、支持拷贝数变异的数据合并,可以用来直接跑GISTIC 三、优化了临床随访信息中疾病进展状态...
- 24
- 16
- zlf
- 发布于 2018-12-11 14:01
- 阅读 ( 14075 )
零代码系列之三教你如何进行功能富集
上一节课讲解如果使用工具盒中一键式WGCNA软件并且找到共表达模块,那么接下来我们看下如何获取目标模块基因集,已经目标模块的具体有哪些功能。1、功能模块基因获取;2、在线富集分析操作步骤;3、富集结果两种可视化方法。
- 1
- 0
- 调研图
- 发布于 2018-12-10 16:14
- 阅读 ( 6040 )
零代码系列之三教你如何看懂WGCNA分析结果
上一节课已经成体系的讲解数据下载到处理完成完整过程,本节可开始讲解如何一键式WGCNA工具使用。1、WGCNA方法总结;2、一键式WGCNA操作说明;3、结果的解读以及参数的调试;4、目标模块分析。
- 2
- 3
- 调研图
- 发布于 2018-12-10 15:54
- 阅读 ( 20488 )