新出炉的Cytoscape视频教程
Cytoscape已成为网络图绘制的核心工具,基因表达调控网络、蛋白互作网络、miRNA-gene调节关系、分析流程、组织架构等任何与网络、结构、层级有关系的事情都可以用Cytoscape来绘制。前期的教程
- 0
- 0
- shengxinbaodian
- 发布于 2018-08-27 20:24
- 阅读 ( 7233 )
文章分享(六)膀胱癌特异性基因组改变,M2可能是巨噬细胞浸润的重要驱动因素?
作者从多个角度研究了膀胱癌中巨噬细胞浸润,特别是M2巨噬细胞,联系了肿瘤微环境与基因组改变,研究的角度方法值得借鉴,感兴趣的小伙伴可以仔细研读。
- 2
- 0
- 柚子
- 发布于 2019-07-25 15:33
- 阅读 ( 7215 )
基因表达与甲基化表达谱纯生信分析4分文章,你也可以
好久没有写稿子了,今天分享一篇纯生信文章,发表在scientific reports上,虽说很多人吐槽SR为水刊,个人认为其实人家发表的很多文章还是很值得学习的,比较喜欢直接一点,我们看图说话哈。...
- 7
- 1
- 余繁荣
- 发布于 2019-03-21 12:26
- 阅读 ( 7214 )
宏基因组扩增子最新分析流程QIIME2:官方中文帮助文档
QIIME2是微生物组分析流程QIIME(截止17.9.10被引8335次)的全新版(不是升级版),采用python3全新编写,并于2018年1月全面接档QIIME,是代表末来的分析方法标准(大牛们制定方法标准,我们跟着用就好了)
- 2
- 5
- 刘永鑫
- 发布于 2017-10-13 20:09
- 阅读 ( 7187 )
微生物组:3分和30分文章差距在哪里?
好的分析和可视化,可以提供大量的信息,同时兼顾简洁优雅。 今天我们抛开实验设计、方法和工作量等因素,仅从文章最吸引人的图片来讨论3分和30分(顶级)文章差距在哪里?
- 0
- 4
- 刘永鑫
- 发布于 2018-01-14 21:31
- 阅读 ( 7182 )
Nature:地球微生物组计划首发成果
Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J,Locey KJ et al (2017). A communal catalogue reveals Earth’s multiscalemicrobial diversity. Nature. 本文今年11月1日在线发表,23...
- 0
- 3
- 刘永鑫
- 发布于 2017-11-28 15:24
- 阅读 ( 7179 )
文献解读(4)NC甲状腺癌的刻画
在这项研究中,作者使用不同类型的测序技术,刻画了ATC和DTCs的基因组和转录组景观。使用突变谱证实了之前的描述,多次基因改变促进了TC的进展。此外,作者采用多种分析方法刻画了TCs转录组特征。
- 0
- 0
- 柚子
- 发布于 2019-12-13 17:37
- 阅读 ( 7156 )
孟德尔随机化之因果推断的假设(二)
3.2 查找有效的工具变量 工具变量(IV )技术是可用于估算因果效应的几种方法之一,而无需完全了解所有可能影响暴露- 结局??
- 0
- 0
- 米老鼠
- 发布于 2020-09-13 23:22
- 阅读 ( 7143 )
illumina的bead 系列表达芯片扫盲
表达芯片大家最熟悉的当然是affymetrix系列芯片啦,而且分析套路很简单,直接用R的affy包,就可以把cel文件经过RMA或者MAS5方法得到表达矩阵。illumina出厂的芯片略微有点不一样,它的原始数据...
- 1
- 1
- 生信菜鸟团
- 发布于 2017-08-10 22:58
- 阅读 ( 7135 )
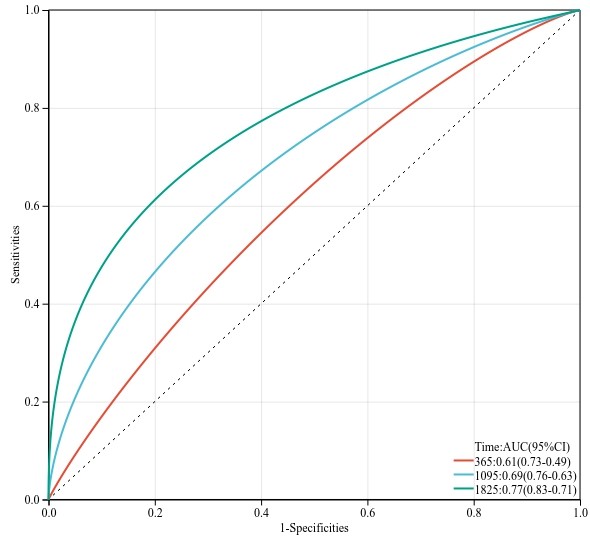
ROC简易分析工具使用
ROC分析主要针对 存在一组真实的连续型数值数据设定阈值的不同对响应变量(二分类)的影响(真阳性率、假阳性率),它常用于 模型的构建过程中对结果预测的性能的一个评估,我们主要分为两类...
- 0
- 0
- admin
- 发布于 2021-09-15 15:07
- 阅读 ( 7109 )
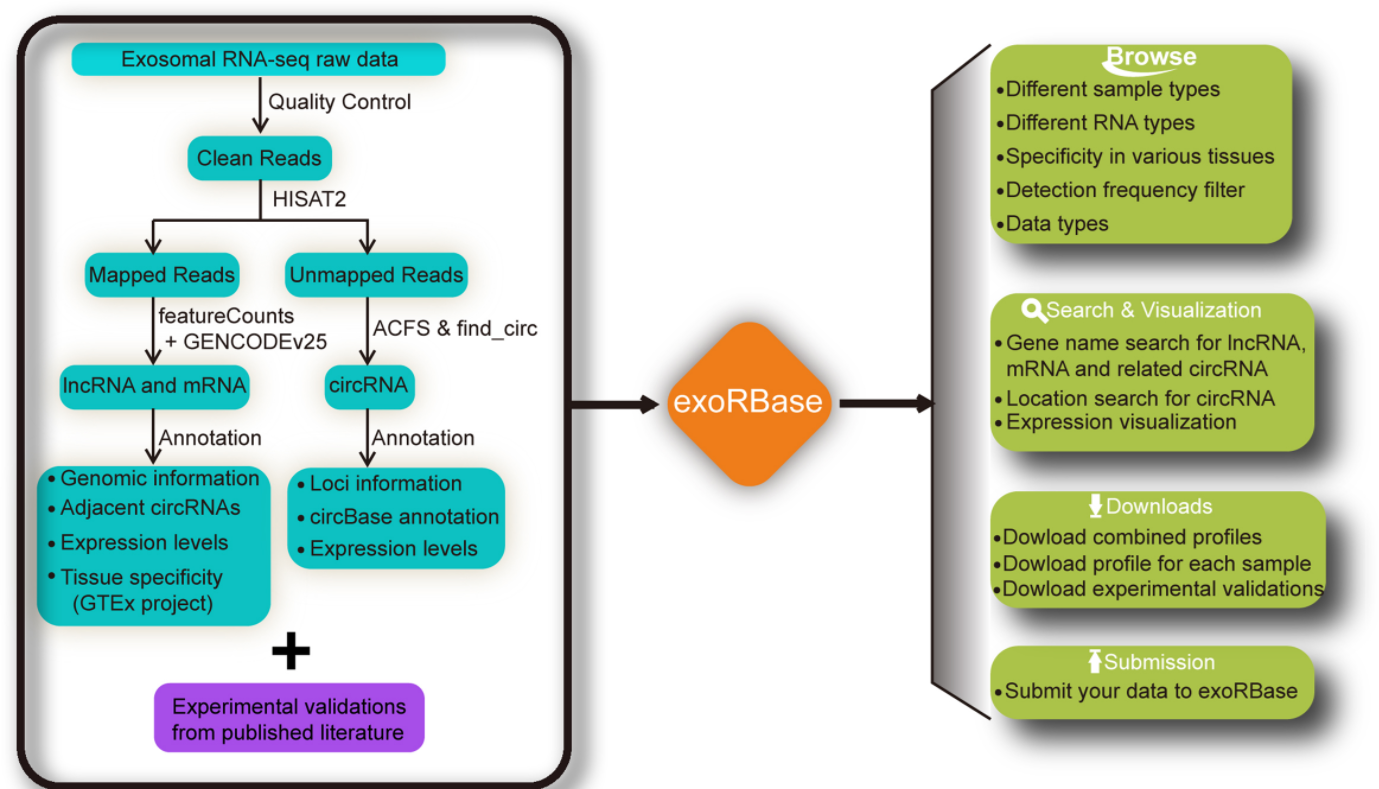
exoRBase: a database of circRNA, lncRNA and mRNA in human blood exosomes
exoRBase一个整合血液外泌体RNA的数据库。
- 1
- 4
- 张海伦
- 发布于 2017-10-13 09:51
- 阅读 ( 7063 )
ConsensusClusterPlus未选择最优阈值的用法案例
我们常用ConsensusCluster来做聚类或者寻找分子亚型,常见的用法是考虑CDF下降坡度稳定小,比如图B、D中应该是k=5,然而有时候选择该值未必完全遵循此规律。 比如这篇CCR的文章,就没有按照...
- 0
- 0
- zlf
- 发布于 2018-10-08 09:10
- 阅读 ( 7003 )

使用sigclust来评估你的聚类是否显著
看到文献中(doi:10.1080/2162402X.2017.1392427)有人使用sigclust来评估聚类是否合理,于是乎研究了这个包。 sigclust主要针对生物信息学数据(高维低样本)产生的聚类结果进行评估,一个基本假...
- 1
- 1
- zlf
- 发布于 2019-02-23 14:36
- 阅读 ( 6927 )
Nat. Biotechnol.扩增子测序革命—用16S及18S rRNA全长进行微生物多样性研究
本文“宏基因组”公众号原创。 作者:舟行天下编辑:metagenome 摘要 前段时间热心肠先生导读了《Nature子刊:高通量&无偏差,分析微生物群落的新方法》。 文中摘要提到:1.几十年以来细...
- 0
- 1
- 刘永鑫
- 发布于 2018-02-10 22:41
- 阅读 ( 6911 )
存在cdf文件的芯片数据,但没有R包构建R包并安装cdf
关键命令: BiocManager::install('makecdfenv') library(makecdfenv) make.cdf.package("GPL15048_HuRSTA_2a520709.CDF.gz", "hursta2a520709cdf", species = "Homo sapiens", compress = T...
- 0
- 1
- zlf
- 发布于 2020-09-04 20:28
- 阅读 ( 6892 )