辉骏生物—串联亲和纯化/质谱(Tandem affinity purification,TAP/MS)
服务简介TAP/MS是Co-IP/MS的“近亲”,两者在寻找诱饵蛋白的互作蛋白以及纯化互作蛋白的原理方面非常相似。但是TAP/MS经过2步亲和纯化,而Co-IP/MS只有1步亲和纯化,故而TAP/MS可以有效减少非特异...
- 0
- 1
- 辉骏生物
- 发布于 2018-11-20 16:25
- 阅读 ( 7570 )
R包装不上怎么办?不用怕分分钟教你如何搞定
最近小编发现大家再用小工具时候总是有一些R包安装不上,今天小编交给大家一个方法。这个是很多免安装的R包提供给大家,欢迎大家使用
- 1
- 0
- 调研图
- 发布于 2018-09-10 17:20
- 阅读 ( 7556 )
他山之石可以攻玉:又能发SCI,毕业了!
《诗经》里有一句话一直广为流传。“他山之石,可以攻玉”,意思是别的山头的石头坚硬,可以琢磨玉器。 作为一只科研汪,在科研上,我们同样可以借用“别人山头的石头”,来琢磨“
- 10
- 0
- 余繁荣
- 发布于 2018-11-21 13:26
- 阅读 ( 7529 )
扩增子分析还聚OTU就真OUT了,试试unoise3
宏基因组领域是当今热门领域,也正是方法快速发展和变革的时代。之前还把 97%聚类OTU作为扩增子行业的金标准。转眼间各位大佬纷纷向OTU聚类方法拍砖,都不建议再使用。 Feature代替OTU是趋势...
- 2
- 4
- 刘永鑫
- 发布于 2017-10-12 23:00
- 阅读 ( 7520 )
实战系列(一)手把手复现3分lncRNA经典小文章
DOI : 10.1080/21691401.2017.1366334 17年发表的与肺鳞癌预后相关的lncRNA研究,影响因子3.026 此篇相对有点早,但是作为一个标准,如果不能完成这样的一个研究,想做近期热点的免疫研究是很难
- 2
- 1
- 生信分析流
- 发布于 2019-07-10 09:47
- 阅读 ( 7515 )

分边小提琴图绘制工具
分边小提琴图(Split violin plot)类似于分组小提琴图,但更便于进行两两(类似癌与癌旁、处理和对照、预后差和好等)比较,如下图 本工具旨在快速简易的绘制分边小提琴图,采用的是 长数据...
- 0
- 1
- admin
- 发布于 2021-06-28 17:08
- 阅读 ( 7408 )
做肿瘤学研究,这些常见的术语你知道吗?
Oncologists — doctors who treat cancer — use many terms when they talk about this disease. These terms usually have very specific meanings that are important in cancer research, specifically in clinical trials. Your doctor may also use these ter
- 1
- 0
- 生信分析流
- 发布于 2018-08-16 13:43
- 阅读 ( 7345 )
服务器里删过SSL的后遗症,R不能正常的安装包了,错误:status was 'Peer certificate cannot be authenticated with given CA certificates
技术不小心删了openssl,然后没挽救回来,通过各种折腾终于能进系统了,各方便都感觉修复了,唯独这个R无法正常的使用install.packages和biocLite来安装依赖包,究其原因则是CA证书错误,status...
- 0
- 1
- zlf
- 发布于 2018-10-11 11:18
- 阅读 ( 7333 )
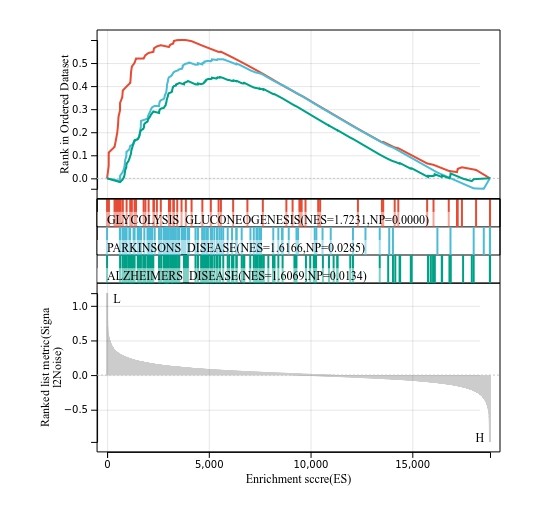
GSEA简易分析及可视化工具
Gene Set Enrichment Analysis (基因集富集分析,GSEA)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知...
- 0
- 2
- admin
- 发布于 2021-08-04 20:12
- 阅读 ( 7332 )
比PCA更好用的监督排序—LDA分析、作图及添加置信-ggord
线性判别分析LDA 线性判别分析,英文Linear Discriminant Analysis, 以下简称LDA。LDA在模式识别领域(比如人脸识别,舰艇识别等图形图像识别领域)中有非常广泛的应用,在生物学大数据研究中...
- 1
- 2
- 刘永鑫
- 发布于 2018-01-07 09:16
- 阅读 ( 7328 )
要研究细胞系,基因?你可能会对这个网站爱不释手哦
细胞系是很多基础研究的同学主要的研究的对象,也是体外实验不可缺少的步骤,我们往往会想知道 某个基因在某个细胞系的表达情况 ,为啥呢? 因为如果该细胞有某种表型B比如增殖能力
- 0
- 0
- 生信分析流
- 发布于 2018-08-23 15:49
- 阅读 ( 7316 )
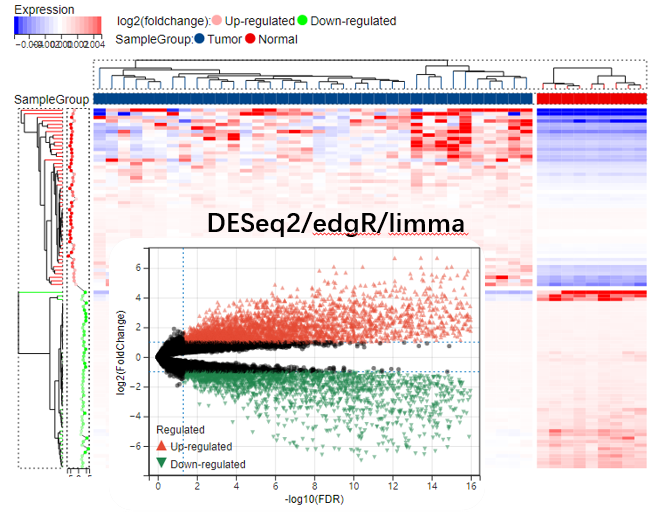
转录组Count数据差异分析工具
当我们拿到表达谱数据之后,做差异分析之前,有两个关键信息需要确认,一个是 数据类型(芯片数据、FPKM、TPM、Count),一个是 哪些样本作为参照物,哪些样本作为 处理组样本,也就是 谁相对于...
- 0
- 3
- admin
- 发布于 2021-08-19 11:40
- 阅读 ( 7315 )
文献解读(1)用免疫风险评分预测黑色素瘤转移的风险
发表期刊:Front Bioeng Biotechnol 发表日期:2020 Mar 31 影响因子:5.12 DOI:10.3389/fbioe.2020.00206
- 0
- 1
- 柚子
- 发布于 2020-06-15 15:37
- 阅读 ( 7308 )
人类lncRNA 简介
Part 1.目前研究相对比较透彻的lncRNA汇总 与癌症相关lncRNA lncRNA长度Cytoband癌症类型HOTAIR2158nt12q13.13乳腺癌MALAT1/a/NEAT27.5 kb11q13.1乳腺癌、肺癌、子宫癌、胰腺癌、宫颈癌、前...
- 3
- 2
- 生信分析流
- 发布于 2018-09-13 15:37
- 阅读 ( 7246 )
你想要的宏基因组-微生物组知识全在这(180701)
宏基因组/微生物组是当今世界科研最热门的研究领域之一,为加强本领域的技术交流与传播,推动中国微生物组计划发展,中科院青年科研人员创立“宏基因组”公众号,目标为打造本领域纯干货技术及思想交流平台。
- 2
- 18
- 刘永鑫
- 发布于 2017-10-31 20:15
- 阅读 ( 7238 )